Introduzione
La leucemia a cellule capellute (HCL) è una neoplasia linfoide a cellule B. L’HCL si differenzia da altre neoplasie a cellule B a causa delle sue caratteristiche morfologiche, immunofenotipiche e molecolari. La sua caratteristica principale è l’accumulo di cellule B monoclonali, con proiezioni simili a capelli sulla sua superficie, presenti principalmente nel sangue periferico, nel midollo osseo e nella milza. È stato descritto nel 1958 da Bournoncle et al. Tuttavia, l’hanno chiamata reticoloendoteliosi leucemica.,1 Il termine “cellule pelose” è stato coniato nel 1996 da Schrek et al.2 Oggi è classificato dall’Organizzazione Mondiale della Sanità come linfoma non Hodgkin a cellule B.3
Definizione
L’HCL è un disturbo raro, cronico, linfoproliferativo delle cellule B. Le sue caratteristiche principali sono i prolungamenti citoplasmatici che conferiscono alle cellule un aspetto peloso.
Eziologia
Sebbene ci siano diversi studi che mostrano un legame tra l’esposizione a determinati agenti e l’HCL, la sua eziologia non è stata chiaramente stabilita, il che potrebbe essere spiegato a causa della sua bassa incidenza., Tra gli agenti che hanno dimostrato un legame positivo ci sono: esposizione a pesticidi, 4 erbicidi, oli minerali, lavoro come falegname o agricoltore.4-7 Recentemente, è stato descritto un legame positivo con le dimensioni,5 mentre il fumo ha un’associazione inversa, specialmente negli uomini.8 Il meccanismo specifico che conferisce la protezione non è noto; tuttavia, è stato suggerito che il fumo diminuisca la gravità dei meccanismi infiammatori9 e che la nicotina induca l’apoptosi nei linfociti.10
Epidemiologia
HCL rappresenta il 2-3% di tutte le leucemie. Ci sono 600 nuovi casi segnalati all’anno negli Stati Uniti (3.,2 casi per milione di abitanti). L’età media alla diagnosi è di 52 anni ed è più comune negli uomini che nelle donne, con un rapporto 4:1; con una maggiore incidenza nella popolazione bianca, specialmente tra gli ebrei ashkenaziti.3 In Messico, l’HCL rappresenta l ‘ 1,12% di tutte le leucemie. Tuttavia, nel nord del paese rappresenta fino all ‘ 1,83%, simile alle informazioni degli Stati Uniti 11, 12
Fisiopatologia
L’HCL è un disturbo cronico linfoproliferativo delle cellule B. Tuttavia, le sue cellule non hanno l’aspetto di alcuna sub-popolazione di cellule B e la sua origine è stata oggetto di dibattito., L’analisi dei geni delle regioni variabili delle immunoglobuline (Ig) è uno strumento utilizzato per scoprire l’origine clonale delle cellule linfoidi.13 In oltre l ‘ 85% dei casi, siamo in grado di trovare mutazioni somatiche nei geni delle regioni variabili Ig delle cellule HCL,14,15 che è un’indicazione che le cellule coinvolte sono passate attraverso il centro germinale o gli organi periferici linfoidi.16 Circa il 40% delle cellule HCL co-esprimono più isotipi Ig clonalmente correlati.17
L’evidenza suggerisce un centro di origine post germinale nelle cellule B di memoria, a causa del loro profilo di espressione genomica.,18,19 L’origine delle cellule B della memoria è compatibile con la mancanza di HCL di traslocazione reciproca cromosomica.20 Assenza di CD27 è tipico in HCL. Questo rappresenta un punto contro l’ipotesi della sua origine nelle cellule B della memoria.21 Tuttavia, sono state osservate cellule B di memoria CD27 negative.22 Poiché l’affezione linfonodale è rara, è stato suggerito che la cellula di origine HCL si trova probabilmente nel midollo osseo o nella milza, poiché quelli sono di solito i luoghi più colpiti. Le cellule HCL mostrano un profilo di espressione simile alla zona marginale della milza.,23
L’aspetto caratteristico dell’HCL è dovuto all’espressione di beta-actina, che viene polimerizzata in F-actina, situata nel citoscheletro corticale.La fosfoproteina 24 PP52, che è specifica per i leucociti, è legata alla F-actina ed è responsabile del supporto alle proiezioni simili ai capelli.25 D’altra parte, il sequenziamento del gene HCL ha recentemente identificato la presenza di una mutazione BRAF V600E in quasi tutti i pazienti con la malattia, assente in altre neoplasie linfoidi a cellule B.26,27 mutazioni BRAF attivano la via MAPK, promuovendo crescita, sopravvivenza e differenziazione cellulare HCL.,28
Presentazione clinica e risultati di laboratorio
Il decorso clinico della malattia è indolente. La maggior parte dei pazienti di solito presenta debolezza e affaticamento come sintomi predominanti durante l’insorgenza della malattia.29 A volte, c’è una storia di infezioni ripetute. I risultati dell’esame fisico sono: splenomegalia nel 96%, epatomegalia nel 58% e linfoadenopatia solo nel 35%. Questi linfonodi ingrossati sono raramente osservati nella periferia; tuttavia, sono generalmente presenti nell’addome e rilevati da studi di imaging.,30
In uno stadio avanzato della malattia siamo in grado di trovare dolore nel quadrante superiore sinistro, infezioni, febbre ed emorragie e/o perdita di peso. Tuttavia, questo è raro a causa della disponibilità e dell’efficacia del trattamento.30,31 Le manifestazioni cliniche sono il prodotto dell’accumulo di cellule capellute nella milza, nel fegato e nel midollo osseo (Tabella 1).32
Manifestazioni cliniche.,
| Milza | Fegato | midollo Osseo | Linfonodi |
|---|---|---|---|
| Le cellule si accumulano nella polpa rossa e atrofia della polpa bianca. Successivamente formano i cosiddetti “pseudo-sinusoidi” attraverso la sostituzione delle cellule endoteliali della polpa rossa da canali vascolari potenziati, contribuendo all’anemia. | Qui, si accumulano nelle sinusoidi epatiche e nel tratto portale., C’è fibrosi in quest’ultimo a causa dell’abbondante acido ialuronico, che stimola le cellule capellute a produrre fibronectina, con la sua corrispondente fibrosi. | In quest’area, c’è un’ampia produzione di fibrosi e soppressione dell’emopoiesi. Il fattore chiave è l’interazione delle cellule capellute con l’acido ialuronico della matrice extracellulare, generando fattori di crescita fibroblastici (FGF) e stimolando le cellule maligne a produrre e secernere fibronectina. | Generalmente, assente della malattia. Mancanza di recettori per l’ingresso di cellule capellute., |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosis
HCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v)., L’ultimo si verifica nel 10% dei casi con un’età media di 70 anni e, nonostante le somiglianze con la classica leucemia a cellule capellute, divergono in assenza dei marcatori immunofenotipici CD25 e CD123. Un altro modo di fare una diagnosi differenziale è la mancanza di risposta al trattamento HCL standard e l’inesistenza di mutazioni del gene BRAF V600F.30
Metodi diagnostici
La diagnosi HCL viene comunemente effettuata con una biopsia e un’aspirazione del midollo osseo combinata con una caratterizzazione immunofenotipica attraverso la citometria a flusso.,33 È importante sottolineare che questa patologia è solitamente sub-diagnosticata e richiede sospetto clinico e l’uso della tecnologia adeguata per risolvere questo problema. Come accennato in precedenza, la maggior parte dei pazienti (70-90%) presenta pancitopenia, con leucopenia (
×109/L), anemia (g/dL), neutropenia (×109/L), monocitopenia (×109/L) e trombocitopenia (×109/L). Solo tra il 10% e il 20% presentano leucocitosi moderata (>10×109/L). I pazienti HCL mostrano elevati livelli sierici di IL-2R (CD25), che si correlano con il grado di attività della malattia.,34 Altri test che dovrebbero essere considerati quando si effettua la diagnosi sono i livelli sierici di immunoglobuline, così come il gene IgVH e le mutazioni somatiche BRAF V600E.32 Alcuni HCL istopatologica e immunofenotipici caratteristiche sono le seguenti:
- •
Linfociti citometria a flusso nel sangue periferico o nel midollo osseo con cellule CD19, CD20, FMC7, CD11c, CD103, CD25, HC2, CD22, sIg, CD79a e CD123 espressioni con le quattro principali e specifici marcatori: CD11c, CD103, CD25 e CD123.34 Comunemente marcatori negativi sono CD5, CD23, CD10, CD79b e CD27.,32
- •
Una forte espressione per CD200 è caratteristica dell’HCL e può essere utile nella diagnosi di casi difficili.
- •
L’aspirazione del midollo osseo con un ago può essere difficile da ottenere ed è spesso improduttiva o”secca”. In una biopsia del midollo osseo, possiamo osservare la fibrosi, con un aspetto cellulare “uovo fritto” causato dagli ampi spazi tra i nuclei e l’abbondante citoplasma. Vengono condotte analisi immunoistochimiche per CD20 e TRAP (fosfatasi acida resistente al tartrato), DBA-4 e annexin A1, che sono caratteristicamente positive.,32
Andrulis et al. ha diretto uno studio in cui è stata riportata l’efficienza dell’anticorpo VE1 per il rilevamento di BRAF V600E, insieme all’identificazione di HCL in altre entità. Inoltre, uno studio condotto da Uppal et al. trovato una sensibilità del 88% e una specificità del 97% per rilevare questa mutazione con l’anticorpo menzionato.34
Trattamento attuale
L’HCL ha regolarmente un’evoluzione indolente. Aspettare e osservare è una buona opzione per i pazienti asintomatici, poiché il trattamento precoce non offre alcun tipo di beneficio nei tassi di sopravvivenza di questi casi., In ogni caso, la progressione della malattia nella maggior parte dei pazienti porterà a complicazioni a causa di citopenie e splenomegalia; cioè anemia, emorragie, infezioni ricorrenti, ecc. Nella pratica generale, il trattamento deve essere iniziato se si verifica uno qualsiasi dei criteri elencati nella Tabella 2.35 Quando viene presa la decisione di non iniziare il trattamento, il monitoraggio clinico e di laboratorio deve essere effettuato ogni tre mesi durante il primo anno e successivamente ogni sei mesi.,35 Il trattamento con HCL non è considerato curativo; ma le attuali strategie di trattamento sono in grado di raggiungere remissioni prolungate, aumentando così i tassi di sopravvivenza globale (Algoritmo 1).36-39
Criteri per iniziare il trattamento.
1. Splenomegalia sintomatica
2. Citopenia che coinvolge almeno uno dei seguenti:
-Emoglobina g/dL
-Piastrine ×109/L
-Neutrofili ×109/L
3., Infezioni gravi
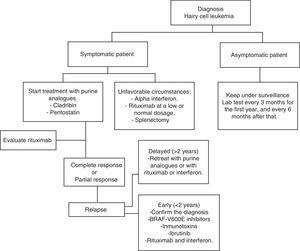
Grafico per la gestione di un paziente con leucemia a cellule capellute.
Analoghi purinici
Nel 1960, i risultati hanno mostrato che il 30% dei bambini con sindrome da immunodeficienza combinata grave mancava dell’enzima deaminasi adenosina (ADA).,40 Inoltre, hanno scoperto che l’accumulo della forma di deossiadenosina trifosfato era responsabile della diminuzione dei linfociti.41 Tenendo presenti queste osservazioni, sono stati sviluppati farmaci in grado di legarsi irreversibilmente ad ADA o di antagonizzare la sua azione. Dopo il trattamento con analoghi delle purine, l’accumulo di desossiadenosina trifosfato provoca la rottura e l’inibizione della riparazione del DNA, che si traduce in apoptosi cellulare.,42
Pentostatina
Noto anche come 2′-deossicoformicina (dcF), un prodotto di Streptomyces antibioticus e inibitori ADA, introdotto per la prima volta nel 1980 come i primi analoghi purinici per il trattamento con HCL. Nella Tabella 3,37,39,43,44 studi specifici mostrano tassi di risposta alla pentostatina. L’uso endovenoso della pentostatina ad un rapporto 4mg/m2 una volta ogni due settimane fino a raggiungere la risposta completa è stato approvato negli Stati Uniti Pentostatina è sicuro in pazienti con alleggerimento della creatinina > 60mL / min. Tuttavia, è necessaria una riduzione del dosaggio se questa depurazione è compresa tra 40 e 60 ml/min.,32 Si raccomanda l’idratazione del paziente con 1,5 L di soluzione endovenosa ad ogni ciclo di pentostatina.30
La risposta globale varia tra l ‘88% e il 96%, mentre la risposta completa è tra il 44% e l’ 81%. Flinn et al. stimato un tasso di sopravvivenza di 5 e 10 anni del 90% e 81%, rispettivamente, con una durata media di follow – up di 9,3 anni.45 Pentostatina è generalmente ben tollerato e gli effetti avversi più comuni sono anemia, trombocitopenia e neutropenia.,Tuttavia, è stato riportato che la pentostatina riduce significativamente la conta linfocitaria CD4+ e CD8+, il che potrebbe aumentare le neoplasie secondarie e l’incidenza di infezioni.47,48
Cladribina
È noto come 2-clorodeossiadenosina (CdA). Nella Tabella 4,49–52 gli studi indicano la sua efficacia. Lo schema più utilizzato consiste nella somministrazione di 0,1 mg/kg/die in infusione continua per 7 giorni. In uno studio non randomizzato, vi erano prove che dimostravano che non vi era alcuna differenza statisticamente significativa nella risposta e negli intervalli di tossicità tra le infusioni (24h e 2h).,Un altro studio randomizzato ha confrontato la somministrazione giornaliera e settimanale di cladribina; non sono stati riscontrati risultati significativi in termini di risposta, sopravvivenza, tassi globali e tossicità.54 Uno studio diverso ha mostrato che il programma settimanale riduceva il rischio di infezioni.55 Uno dei vantaggi della somministrazione sottocutanea è il fatto che nella maggior parte dei casi non richiede il ricovero in ospedale. 0,14 mg / die per 5 giorni ha un tasso di risposta del 95%, 56 simile alla somministrazione endovenosa. I programmi sottocutanei settimanali hanno tassi di risposta e tossicità simili a quelli giornalieri.,57 Con un solo ciclo di cladribina, è possibile ottenere una risposta globale fino al 100% e le percentuali di risposta totale variano dal 77% al 95%.49-52 Jehn et al. ha riportato una sopravvivenza globale a 12 anni del 79%.In generale, la cladribina è ben tollerata, con citopenie e febbre che sono gli effetti avversi più comuni.
Studi sull’efficacia della cladribina nei casi di HCL.,
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days |
77.3 | 18.,6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days |
88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days |
95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.,1mg/kg/d 7 days |
79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence., Anche così, ci sono studi retrospettivi che dimostrano il fatto che entrambi i farmaci hanno un’efficacia simile, in termini di risposta completa e sopravvivenza libera da malattia.
Altri trattamenti
Gli analoghi purinici rimangono la prima linea di trattamento, ma nuove scoperte riguardanti la fisiopatologia dell’HCL hanno portato alla creazione di farmaci con diversi target terapeutici. Questi farmaci sono in fase di ricerca e alcuni hanno mostrato risultati promettenti.
Rituximab
Poiché l’HCL è un tumore maligno a cellule B, è logico utilizzare un anticorpo monoclonale contro CD20, come rituximab., Impiegato come farmaco autonomo, rituximab può raggiungere tassi di risposta totali del 10-54% nei pazienti con recidiva di HCL, a un dosaggio di 375 mg/m2 una volta alla settimana per 4-8 settimane.58,59 Else et al. sono stati esaminati retrospettivamente 18 pazienti che sono stati trattati con analoghi delle purine in associazione con rituximab come seconda linea di trattamento, dopo essere stati trattati con analoghi delle purine come singoli agenti. Tutti i pazienti hanno risposto, con una percentuale di risposta completa dell ‘ 89%.60
Rituximab a 375 mg/m2 a settimana per 8 settimane come terapia iniziale dopo la somministrazione 5.,6 mg/m2 cladribina tramite infusione endovenosa 2-h per 5 giorni genera una risposta totale del 100%.61 In situazioni speciali o sfavorevoli, 100mg di rituximab a settimana può essere utilizzato per 4-6 settimane. Questo è meno costoso ed è di solito efficace, soprattutto quando è combinato con interferone.
Mentre gli analoghi delle purine potrebbero non essere in grado di eliminare l’HCL, poiché la malattia residua minima (MRD) rilevata dopo la somministrazione di cladribina è sempre fortemente CD20+, l’eradicazione della MRD può essere ottenuta utilizzando rituximab. Rivandi et al., dimostrato in uno studio preliminare che rituximab a dosi convenzionali per un periodo di 8 settimane svolge con grande attività, eliminando MRD in 13 pazienti, quando viene utilizzato 4 settimane dopo la somministrazione di cladribina.62
Vemurafenib
Come descritto in precedenza, la mutazione BRAF V600E è la chiave genetica dell’HCL. Pertanto, è un obiettivo terapeutico che è stato studiato negli ultimi anni. Vemurafenib è un inibitore orale di BRAF V600E. Tiacci et al., condotto uno studio per misurare l’attività e la sicurezza di Vemurafenib in pazienti con HLC recidivanti dopo trattamento con analoghi delle purine o refrattari alla somministrazione di analoghi delle purine. Il tasso di risposta globale è stato del 96% e il tasso di risposta completa è stato del 35%, con una sopravvivenza libera da recidive media di 19 mesi. Gli effetti avversi sono stati rash, artralgia e artrite.Poiché è stata osservata una relazione positiva tra l ‘uso di Vemurafenib e l’ insorgenza di neoplasie dermatologiche, si raccomandano frequenti esplorazioni della pelle.,
Ibrutinib
Un inibitore selettivo e irreversibile della tirosina chinasi di Burton interviene nella via di segnalazione delle cellule B.È stato recentemente iniziato un test clinico ibrutinib in pazienti con recidiva di HCL. I dati preliminari di efficacia e sicurezza mostrano effetti avversi come eruzioni, diarrea e artralgia. Questo test clinico è attualmente in corso in diversi centri negli Stati Uniti (NCT01841723).
Immunotossine
Al fine di aumentare la citotossicità degli anticorpi monoclonali, sono state create tecniche che facilitano la produzione di coniugati anticorpo–tossina o anticorpo–farmaco., Un’immunotossina è la fusione tra una tossina batterica (es. Pseudomonas exotoxin o difterite) e la frazione variabile di un anticorpo monoclonale, il cui bersaglio specifico si trova sulla superficie di cellule neoplastiche come CD25 o CD22. Questa tossina viene rilasciata all’interno della cellula neoplastica e interferisce con la sintesi proteica.65
BL22 è un’immunotossina contro CD22 fusa con una forma troncata della P. esotossina PE38. In un test clinico di fase II, BL22 è stato testato in 36 casi di recidiva di HCL o di malattia refrattaria., Dopo un ciclo (40 mg/kg ogni due giorni, tre dosi), il tasso di risposta completa era del 25% e il tasso di risposta globale era del 50%. Queste risposte sono migliorate fino a raggiungere un tasso di risposta totale del 47% e un tasso di risposta globale del 72% dopo ritrattamento (solo in pazienti con citopenie). Due pazienti hanno sviluppato la sindrome emolitica uremica senza la necessità di ricorrere alla plasmaferesi.Successivamente, moxetumomab pasudotox è stato sviluppato come una versione modificata di BL22 con una maggiore affinità e citotossicità., In un test di fase I, che comprendeva 28 pazienti con recidiva e resistenza all’HCL, è stata ottenuta una percentuale di risposta globale dell ‘ 86%, inclusa una percentuale di risposta completa sostenuta nel 46% dei pazienti.67
Opzioni terapeutiche in circostanze sfavorevoli
Anche se l’HCL viene trattato nella maggior parte dei paesi sviluppati con cladribina e pentostatina, è un fatto che questi farmaci non sono solo costosi, non sono disponibili in Messico e in molti paesi con risorse limitate., Quindi, in questi tipi di circostanze, ci sono altre opzioni terapeutiche convenienti con risultati favorevoli
L’interferone alfa per il trattamento del paziente HCL è stato introdotto per la prima volta nel 1984. Oggi il suo uso è limitato, principalmente a causa della grande efficacia degli analoghi delle purine. D’altra parte, nei paesi con scarse risorse economiche, è un’opzione economica e che ha dimostrato risultati simili a quelli della cladribina in termini di sopravvivenza globale. Il suo nome è Ru, condotto uno studio comparativo tra interferone alfa (n=18) e cladribina (n=11), dove la differenza nella sopravvivenza globale tra entrambi i gruppi non era statisticamente significativa; 94% a 217 mesi nel gruppo interferone e 91% a 133 mesi nel gruppo cladribina.68 In uno studio condotto nel nostro centro, nove pazienti HCL hanno ricevuto tre mega-unità IFN tre volte alla settimana per 12 settimane, successivamente hanno ricevuto il trattamento ancora una volta per 8 settimane quando c’è stata una riattivazione della leucemia o dopo 10 mesi di osservazione ogni anno., Tutti i pazienti hanno avuto una remissione ematologica prima di 12 settimane di trattamento. Questa opzione terapeutica è più economica, efficace e paragonabile ad altre forme di terapia con IFN nel trattamento e nel mantenimento di pazienti con questo tipo di leucemia.69 È possibile combinare l’interferone con rituximab senza aumentare gli effetti tossici e migliorare l’efficacia.
La splenectomia è stato il primo intervento che ha modificato significativamente la sopravvivenza nei pazienti. Oggi è usato raramente., Può essere raccomandato in pazienti con splenomegalia massiva dolorosa (>10 cm sotto il bordo costale) e con infiltrazione minima del midollo osseo, o in pazienti refrattari al trattamento con interferone e analoghi delle purine.33 Studi retrospettivi mostrano un tasso di risposta completa del 40-62% e un tasso medio di sopravvivenza a 5 anni fino al 68%.70,71 Lad et al. pubblicato uno studio retrospettivo, tra cui 24 pazienti con diagnosi di HCL, sono stati divisi in due gruppi: 17 pazienti hanno ricevuto cladribina e 7 sono stati splenectomizzati., il 75% dei pazienti nel gruppo splenectomia ha mostrato remissione totale, il 94% lo ha fatto nel gruppo cladribina. Una scoperta interessante quando si confrontano entrambi i gruppi è che non sono state osservate differenze statisticamente significative per quanto riguarda la sopravvivenza libera da leucemia e la sopravvivenza globale.
Prognosi
Il tempo di sopravvivenza nei pazienti dopo la diagnosi era di 4 anni prima che fosse noto un trattamento, a causa di complicazioni derivate dalle citopenie, incluse emorragie e infezioni. Successivamente, con la splenectomia come trattamento di prima linea, c’è stata una risposta completa del 40-62% e un tasso di sopravvivenza di 5 anni al 61-68%., Quindi, l’interferone alfa è stato utilizzato come primo farmaco con benefici nel trattamento dell’HCL. Ancora il suo tasso di risposta completa era basso, al 10%.
Oggi, con analoghi delle purine (pentostatina e cladribina), la risposta completa è indotta fino all ‘ 80% dei pazienti con una sopravvivenza mediana di 10 anni. I tassi di risposta globale sono del 96-100% con un tasso di risposta completo dell ‘80% e un tasso di sopravvivenza di 10 anni compreso tra l’ 85% e il 100%.74 Nonostante ciò, una percentuale significativa di pazienti con HCL fallisce nella risposta al trattamento o diventa resistente., Fino al 48% dei pazienti recidiva nei successivi 15 anni.75 Il futuro dei pazienti HCL è molto favorevole. La sfida è identificare questa neoplasia il prima possibile e trattarla correttamente utilizzando le risorse disponibili.
Conflitto di interessi
Gli autori non hanno conflitti di interesse da dichiarare.