I forrige innlegg har vi introdusert ortho- ,para – og meta – styret i elektrofil aromatisk substitusjon. Tidligere til at vi dekket mekanisme av elektrofil aromatisk substitusjon, og viste at mekanismen går gjennom en carbocation middels.,
i Dag, vi kommer til å knytte de to begrepene sammen, og prøve å vise at om en substituent er en ortho-, para – eller meta – direktør avhenger av hvordan substituent påvirker stabiliteten i carbocation middels.
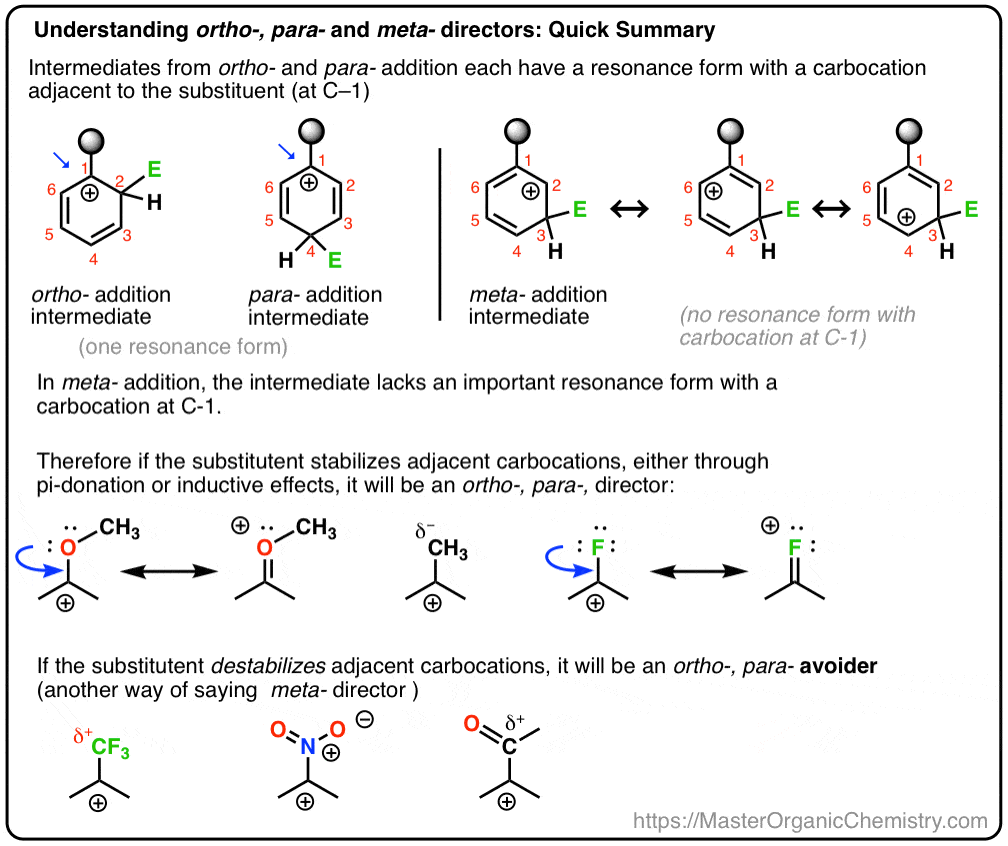
Og vi vil også se hvorfor et alternativt navn for meta – direktør kan bli, «ortho-, para – avoider». : – )
Innholdsfortegnelse
- Carbocations Er Stabilisert Av Tilstøtende Atomer Med Enslig Par
- Case #1: En ortho-,para – Direktør (OCH3)
- Ja, Oksygen Er Electronegative., Men Donasjon Av Sin Enslig Par Mer Enn Gjør Opp For Det!
- Reaksjonen-Energi-Diagram For En Ortho Para Direktør
- Case #2: En meta – direktør (CF3).
- Det Vi Kaller «meta– Styret» Er Mer Som «ortho-, para– Avoiders»
- Reaksjonen Energi-Diagram For En meta – Direktør
- Så Hvorfor Er Halogener Ortho-, Para – Styret?
- Notater
- Quiz deg Selv!,
- (Avansert) Referanser og Videre Lesing
Carbocations Er Stabilisert Av Tilstøtende Atomer Med Enslig Par
La oss tenke tilbake til hvilke faktorer som øker stabiliteten av carbocations.
Carbocations er electron-dårlig arter med seks elektroner i sitt valence shell. Så, noen substituent som kan donere electron tetthet mot carbocation vil være stabiliserende. Dette inkluderer:
- elektron-avgi substituenter som kan fungere som «sigma-givere» (f.eks., mange alkyl grupper, som donerer electron tetthet gjennom induktiv effekter )
- tilstøtende atomer med enslig par som kan fungere som «pi givere» til carbocation
- ved siden C–C pi obligasjoner som kan stabilisere carbocation gjennom lade delocalization («resonans » stabilisering», med andre ord).
Og faktorer som destabilisere carbocations?
- alle substituent som fjerner electron tetthet fra en carbocation, slik som sterke elektron-uttak grupper.
Så her er en rask quiz., Gitt alt som er skrevet ovenfor, hvilken av de to carbocations nedenfor vil være mer stabil?
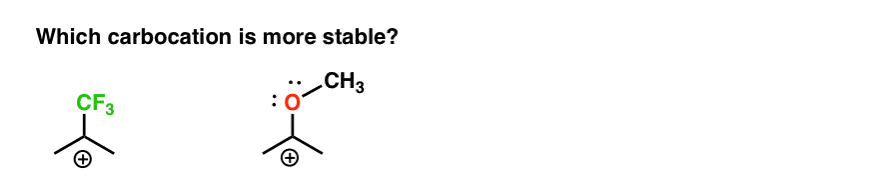
Hvis du kan svare på dette spørsmålet også, du er det meste av veien mot å forstå forskjellen mellom ortho-, para – og meta – styret.
La oss se på de to case-studier. Vi vil se på en generisk elektrofil aromatisk substitusjon reaksjon av benzen med en ortho-, para – direktør (methoxybenzene) og undersøke mellomprodukter som er mottatt fra angrep på ortho, meta og para posisjoner., Deretter vil vi utføre den samme øvelsen med en meta– direktør (trifluoromethylbenzene).
I prosessen, vil vi håper å forstå hvorfor methoxybenzene favoriserer ortho – og para– produkter mens trifluoromethylbenzene favoriserer meta – produkt.
– Case #1: En ortho-,para – Direktør (OCH3)
Elektrofil aromatisk substitusjon av methoxybenzene har en tendens til å gi ortho – og para – produkter med svært lite meta- ., Her er hva som skjer i nitration, for eksempel:
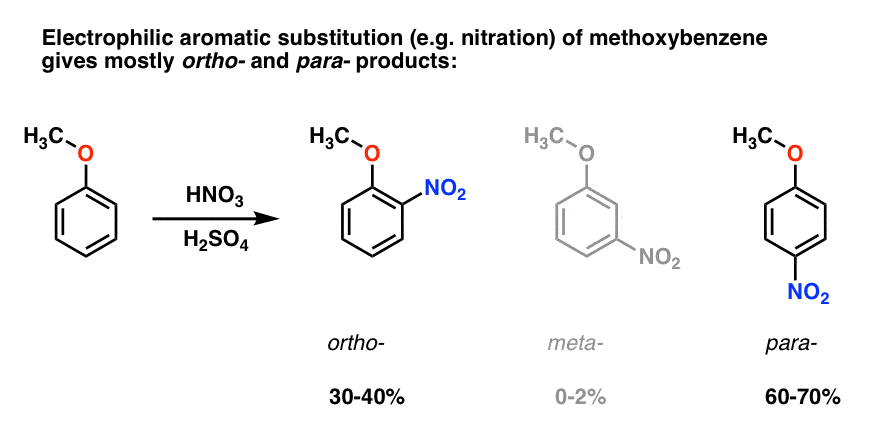
para – produkt dominerer (60-70%), med ortho – tett bak (30-40%), og bare et spor av meta– .
Hvorfor?
3. Ja, Oksygen Er Electronegative. Men Donasjon Av Sin Enslig Par Mer Enn Gjør Opp For Det!
Her er den vei som fører til ortho – produkt., Vi bryte en C–C pi bånd og danne en ny C–E bond (der E er ment å representere en generisk electrophile), genererer en resonans-stabilisert carbocation:
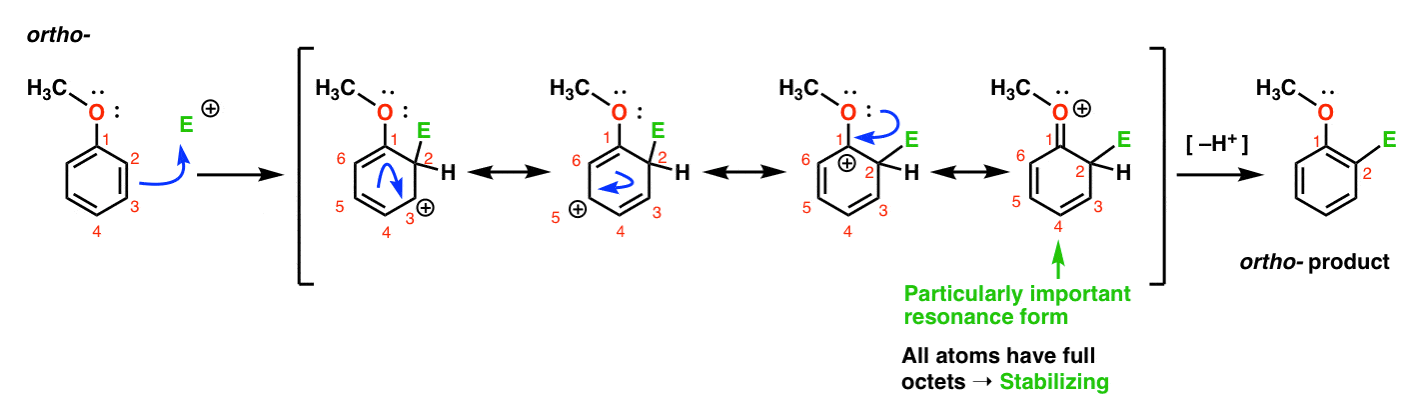
Hvis du ser nøye, bør du være oppmerksom på at vi kan danne fire resonans former . Tre av dem bære det fulle carbocations (på C1, C3 og C5), men det er den fjerde – resonans skjema hvor det er C=O pi bond – noe som er spesielt viktig. Hvorfor? I denne resonansen form, alle karbonatomene har en full oktetten av elektroner., Det er fordi oksygen direkte festet til ringen kan donere en enslig par til tilstøtende carbocation, danner en pi bond. Dette er et eksempel på pi donasjon.
Her er veien for angrep av electrophile på meta – posisjon. Merk forskjellen!
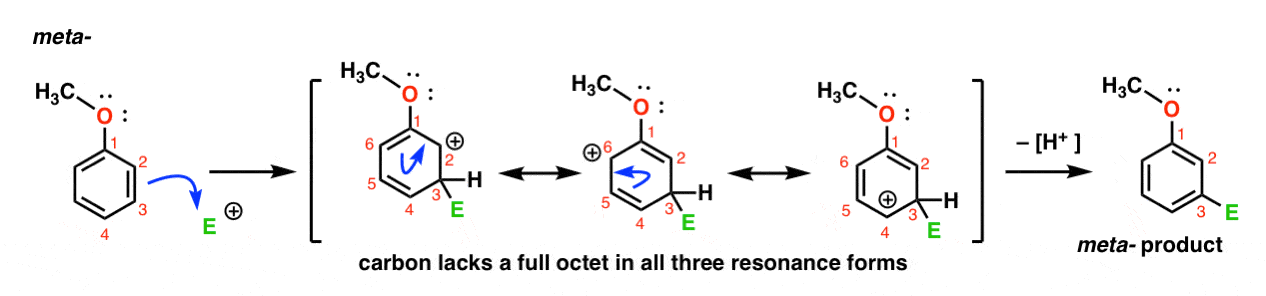
Angrep av electrophile på C-3: resultater i en carbocation som kan være delocalized via resonans til C2, C4 og C6. Men, det er ingen måte å «flytte» den carbocation til C1, noe som betyr at det er ingen rimelig resonans skjema der alle atomene har full oktettene.,
Dette gjør meta – carbocation middels mye mindre stabil enn ortho – carbocation middels.
til Slutt, her er den reaksjon som fører til para – produkt:
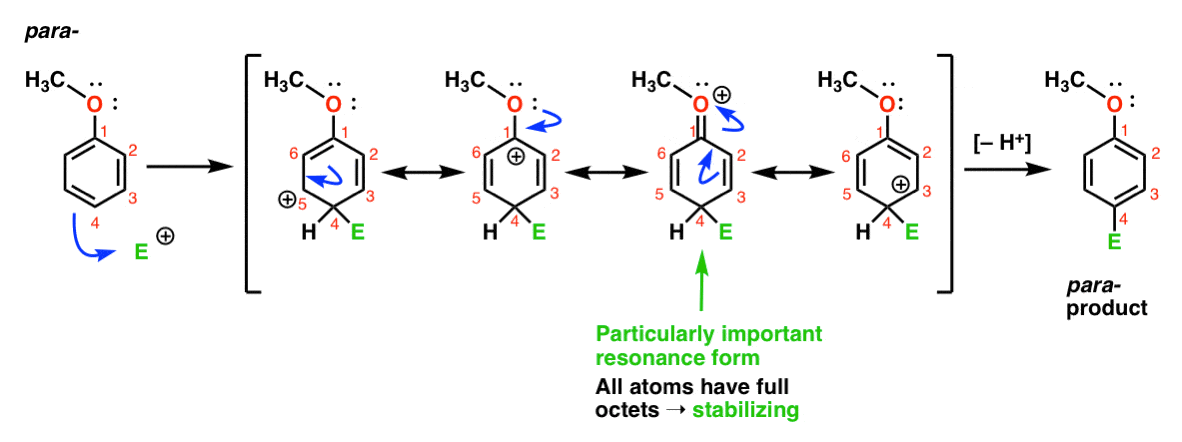
Her kan vi igjen trekke resonans former med carbocations på C1, C3 og C5, samt en fjerde resonans skjema hvor de festet oksygen atom donerer et elektron par til carbocation på C1, noe som resulterer i en full oktett på karbon. Dette er en situasjon i hovedsak de samme som ortho – middels.,
Så, ved analyse av resonans former (og merk mine ord, du kan også bli bedt om å gjøre det samme på en test i nær fremtid) kan vi se at de mellomliggende carbocations som følge av ortho – og para– tillegg er betydelig mer stabile enn de mellomliggende fra meta– tillegg.
Dette forklarer hvorfor er ortho – og para – produkter dominerer, og meta – produkt er mindre.
Det kan være nyttig å visualisere dette ved å tegne en reaksjon energi diagrammet.
4., Reaksjonen-Energi-Diagram For En Ortho Para Direktør
overgangen tilstand som fører til ortho – og para – tillegg produkter er mye lavere energi enn meta– .
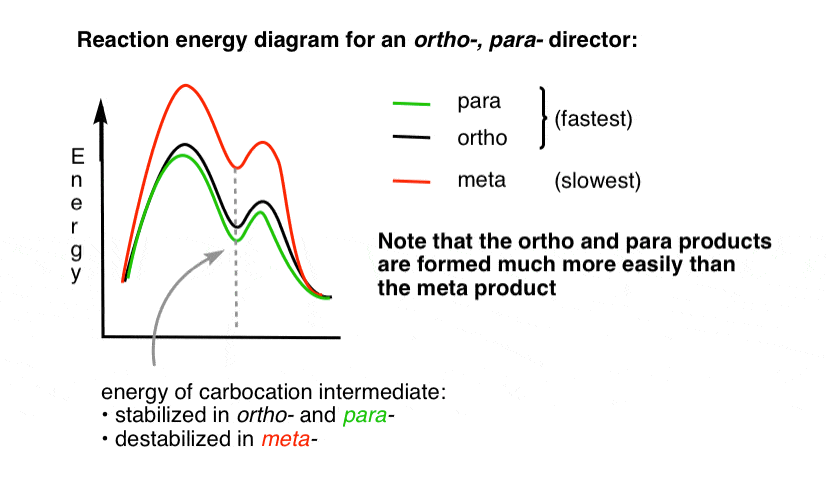
Ett spørsmål – hvorfor tror du at para – kan bli begunstiget (for eksempel 60%-70% avkastning for para– produkt av nitration, ovenfor, mot 30-40% for ortho-) selv om det er to ortho – stillinger og bare en para- ?
Tenk på det et øyeblikk, og vi vil ta det opp nedenfor.
5. Case #2: En meta – direktør (CF3).
Så hva om CF3?, Hvorfor ikke gi meta – produkter? La oss gjøre det samme type analyse.
Her er den vei som fører til ortho – produkt. Som med ortho – middels ovenfor, kan vi trekke tre resonans former å plassere carbocation på C1, C3 og C5, henholdsvis.
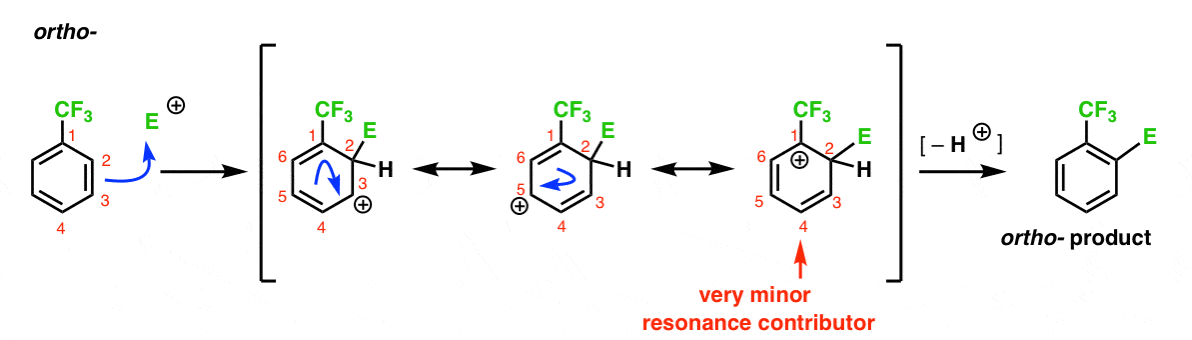
Så hva er annerledes i denne saken?
Hva som er forskjellig, er tilstedeværelsen av en sterk electron uttak av gruppen (CF3) i C1, og dette endrer fullstendig ballgame.
Som vi har sagt, carbocations er destabilized av naboer som trekke elektron-tetthet., Derfor forventer vi at dette skal være en svært liten resonans bidragsyter, med det resultat at den positive ladningen er bare delocalized over C3 og C5.
Vi har nevnt at CF3 er en meta – direktør. Det er rimelig å tenke at det er noe med meta– som gjør det spesielt stabilt. Ved å analysere middels for meta– produkt nedenfor, kan du se hvorfor?
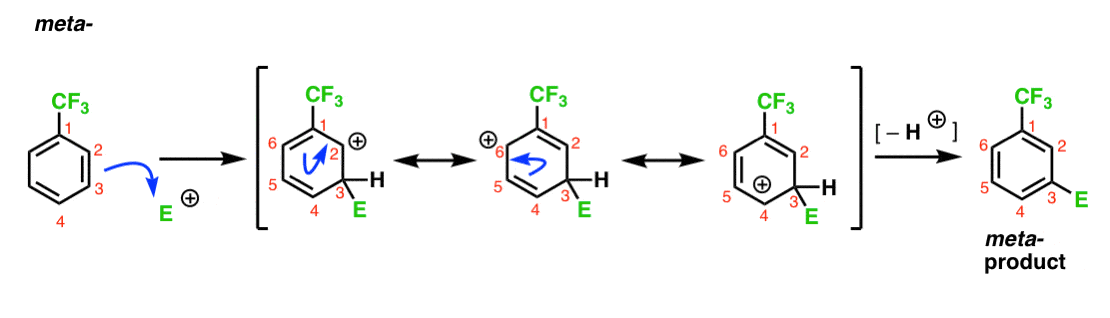
Hmmm. Det er ingen mellomliggende hvor en karbon har en full oktett. Det er ikke spesielt stabilt resonans former.
Det er en slags «meh», er det ikke?
6., Hva Vi Kaller «meta– Styret» Er Mer Som «ortho-, para– Avoiders»
På den annen side, er det ikke noe spesielt ustabil resonans former heller. Siden den positive ladningen er lokalisert på C2, C4 og C6, en situasjon der den positive ladningen er i direkte tilknytning til elektron-uttak CF3-gruppen er unngått. Og den positive ladningen er dermed delocalized pent hele ringen, i motsetning til i ortho – situasjon, som nevnt over.
Så det er en «mindre dårlig» mellomliggende enn ortho-.,
til Slutt, para – mellomliggende har det samme problemet som ortho- ; et mellomliggende med en positiv ladning på C1, i tilknytning til CF3.
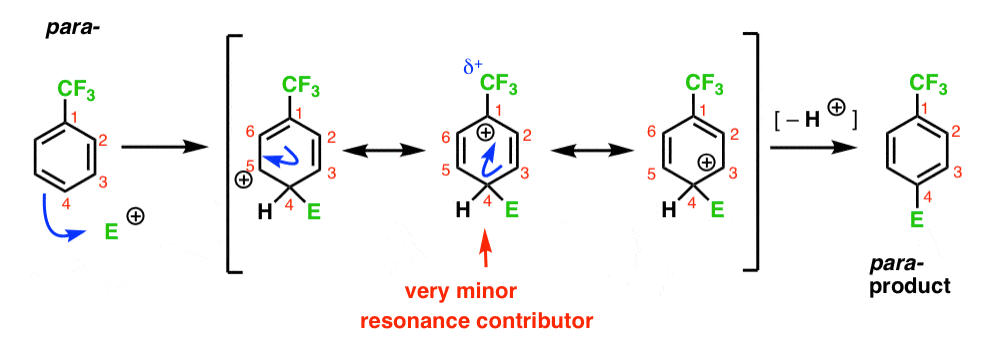
Dette resulterer i bare to viktige resonans former, noe som fører til en mindre delocalized (og derfor mindre stabil) carbocation middels.
Nederste linjen: meta carbocation mellomliggende er mer stabil enn enten ortho– eller para – intermediater.
Men det er ikke fordi substituent seg selv har en stabiliserende effekt på meta– middels. .,
jeg vil argumentere for at det er mer nyttig å tenke på meta – som mindre ustabil enn den ortho – og para– .
På denne måten, det er ikke så mye som CF3 er en meta – direktør, så mye at det er et «ortho– , para– avoider. «
7. Reaksjonen Energi-Diagram For En meta – Direktør
Her er en skisse av reaksjonen energi diagram:
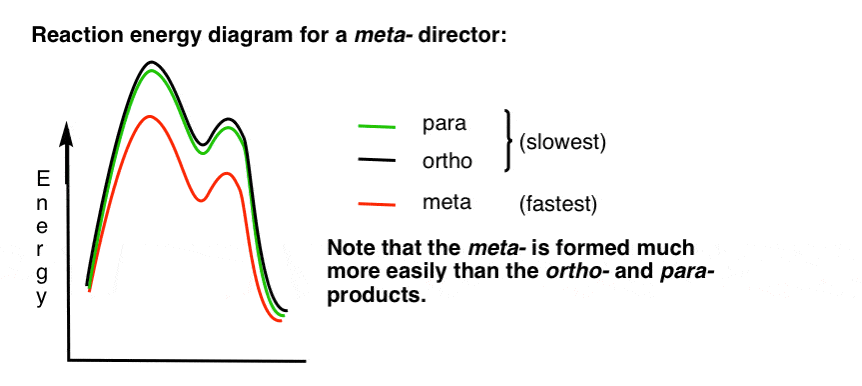
Ok.
Tilbake til vårt opprinnelige spørsmål. Slik som carbocation er mer stabil?,
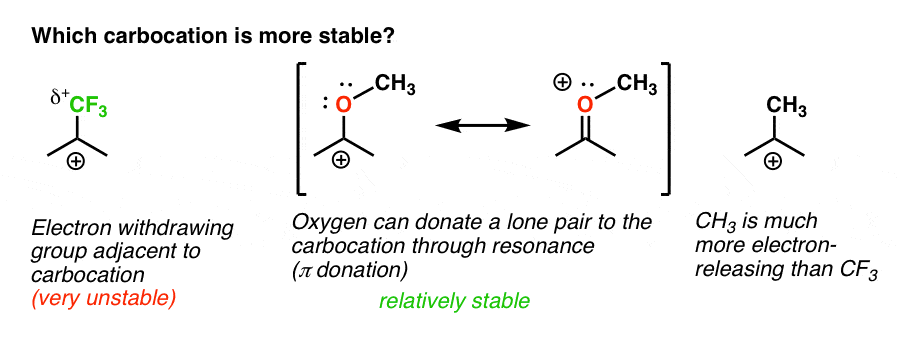
det er Tydelig at carbocation med en tilstøtende oksygen lager en enslig par er langt mer stabil enn en carbocation i tilknytning til et elektron-uttak gruppe som CF3.
Og som består av forskjellen mellom en ortho-, para – direktør som OCH3 og en meta – direktør som CF3.
Det er verdt å merke seg at de fleste alkyl grupper (for eksempel CH3 ) mangler en enslig par, men er fortsatt ortho-, para – styret., Dette er konsistent med alt vi har lært før om hvordan alkyl grupper er generelt stabiliserende for carbocations – husker at carbocation stabilitet øker generelt med substitusjon
Så om de para– produkter…
Hvorfor er para – produkter som er produsert på en større hastighet enn ortho- ?
svaret er steriske hinder.
Angrep på para-posisjon er mindre tynget av substituent enn angrep på ortho – stillinger.
Så Hvorfor Er Halogener Ortho-, Para – Styret?
Dette etterlater oss med noe vanskelig eksempel av halogener.,
Hvorfor er fluor, klor, brom og jod ortho-, para – styret, selv om de er deaktivering av grupper?
svaret bør ikke komme som noen overraskelse hvis du har fulgt med, men vi kommer til å la dette til neste innlegg, fordi dette er lang nok allerede.
Neste innlegg: Hvorfor er halogener ortho-, para – styret?
Notater
Merk 1. Det er mer riktig å si at ortho – og para – produkter dominerer fordi overgangen stater som fører til disse produktene er lavere i energi, snarere enn de energiene av mellomprodukter seg selv., Tross alt, det er energien i overgangen stater som bestemmer aktivering barriere, og derfor reaksjonen pris.
Merk 2. Eller hyperconjugation, som de fleste lærebøker (med unntak av Maitland Jones) generelt unngå.
Quiz deg Selv!,/div>
 Klikk for å Vende
Klikk for å Vende
Klikk for å Vende  Klikk for å Vende
Klikk for å Vende 
(Avansert) Referanser og Videre Lesing
– >
- A., F. Holleman, Die direkte Einführung von Substituenten i den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/recl.19100291205
A. F Holleman fra 1910 sa at ortho–para orientering er forbundet med aktivering og meta retning med deaktivering. - —arten av de vekslende effekt i karbon kjeder. Del XXIII. Avvikende retning av halogener, og dens innvirkning på problemet med ortho–para-forhold, i aromatisk substitusjon
Christopher Kelk Ingold og Charles Cyril Norrey Vass
J. Kem. Soc. 1928, 417-425
TO: 10.,1039/JR9280000417
Dette notatet drøfter regi virkninger i 1,2-dihalobenzenes. - Noen observasjoner om steriske hinder og virkninger av substituenter på ortho : para-forholdet i aromatisk substitusjon
P. B. D. de la Mare
J. Kem. Soc. 1949, 2871-2874
TO: 10.1039/JR9490002871
I dette papiret, de la Mare drøfter ulike faktorer som kan utgjøre en overvekt av para over ortho selektivitet, etter å dele utbyttet av ortho produktet av 2 (siden det er 2 ortho stillinger, men bare 1 para posisjon i monosubstituted benzener)., - Volum virkninger av alkyl grupper i aromatiske forbindelser. Del V. monosulphonation av p-cymene
R. J. W. Le Fèvre
J. Kem. Soc. 1934, 1501-1502
TO: 10.1039/JR9340001501
I p-cymene, den viktigste produkt innhentet på elektrofil sulfonation er 2-produktet (ortho til metyl-gruppen), sannsynligvis på grunn av sterics. - Effekter av Alkyl Grupper i Elektrofil Addisjon og Innbytter
COHN, H., HUGHES, E., JONES, M. og PEELING, M. G.
Natur 1952, 169, 291
TO: 1038/169291a0
Dette papiret har data å sammenligne nitration av t-butylbenzene og toluen., T-butylbenzene er mye mer p-regisserte enn toluen (på 79,5% para for t-butylbenzene vs. 40% para for toluen), som er sannsynlig på grunn av sterics (ortho tilnærming er blokkert av bulkier t-butyl-gruppen). - Distribusjon av Isomerene i Mononitration av Ethyl – og Isopropylbenzene. Ytterligere Bevis for en Steriske Effekt i Isomeren Distribusjon
Herbert C. Brun og W. Hallam Bonner
Journal of American Chemical Society 1954, 76 (2), 605-606
TO: 10.,1021/ja01631a084
Tabell II i denne artikkelen illustrerer at ortho produktet har fått fra nitration av monoalkylbenzenes reduseres som alkyl-gruppen blir større (for eksempel t-butylbenzene gir svært lite ortho produktet ved nitration i forhold til toluen). - —arten av de vekslende effekt i karbon kjeder. Del XXII. Et forsøk på ytterligere å definere den sannsynlige mekanismen for orientering i aromatisk substitusjon
Christopher Kelk Ingold og Firenze Ruth Shaw
J. Kem. Soc. 1927, 2918-2926
TO: 10.,1039/JR9270002918
En tidlig papir av den innflytelsesrike Fysiske Organisk Kjemiker, Prof. C. K. Ingold, sier at halogenobenzenes er induktivt elektron-uttak, men samtidig resonans-stabiliserende. - Avvikende Reaktivitet av Fluorobenzene i Elektrofil Aromatisk Substitusjon og Beslektede Fenomener
Joel Rosenthal og David I., Schuster
Journal of Chemical Utdanning 2003, 80 (6), 679
TO: 1021/ed080p679
En veldig interessant papir, egnet for nysgjerrig undergrads, og drøfter noe som de fleste praktiserer organiske kjemikere vil vite empirisk – fluorobenzene er nesten så reaktive som benzen i EAS eller Friedel-Crafts reaksjoner, som er counterintuitive når en vurderer elektroniske effekter! - Organisk kjemi
N. Haworth, C. K. Ingold, T. A. Henry
Ann. Rep. Prog. Kem. 1926, 23, 74-185
TO: 10.1039/AR9262300074
En tidlig papir diskuterer regi virkninger i EAS. Pp., 136 og 137 inneholde tall som viser flyten av elektroner. Pg. 140 drøfter meta-retning, noe som skjer for ‘motsatt’ grunner som o/p-retning ved alkyl grupper.