introdução
leucemia de células pilosas (HCL) é uma neoplasia linfóide de células B. O HCL diferencia-se de outras neoplasias das células B devido às suas características morfológicas, imunofenotípicas e moleculares. Sua principal característica é a acumulação de células B monoclonais, com projeções Tipo cabelo em sua superfície, principalmente encontrado no sangue periférico, medula óssea e baço. Foi descrita em 1958 por Bournoncle et al. No entanto, chamaram-lhe reticuloendoteliose leucémica.,1 o termo “células cabeludas” foi cunhado em 1996 por Schrek et al.Hoje é classificado pela Organização Mundial de saúde como um linfoma não-Hodgkin de células B.3
definição
HCL é uma doença rara, crónica, linfoproliferativa de células B. Suas principais características são os prolongamentos citoplásmicos que dão às células um aspecto Peludo.etiologia
embora existam estudos diferentes que mostram uma ligação entre a exposição a certos agentes e HCL, a sua etiologia não foi claramente estabelecida, o que pode ser explicado devido à sua baixa incidência., Entre os agentes que provaram uma ligação positiva estão: exposição a pesticidas, 4 herbicidas, óleos minerais, trabalho como carpinteiro ou agricultor.4-7 recentemente,uma ligação positiva com o tamanho tem sido descrita, 5 enquanto que fumar tem uma associação inversa, especialmente em homens.8 desconhece-se o mecanismo específico que confere a protecção; no entanto, sugere-se que o tabagismo diminua a gravidade dos mecanismos inflamatórios 9 e que a nicotina induz a apoptose nos linfócitos.10 Epidemiologia
HCL representa 2-3% de todas as leucemias. Há 600 novos casos relatados por ano nos EUA (3.,2 casos por milhão de habitantes). A Idade Média no diagnóstico é de 52 anos e é mais comum em homens do que em mulheres, com uma proporção de 4:1; com uma maior incidência na população branca, especialmente entre os judeus Ashkenazi.3 no México, HCL representa 1,12% de todas as leucemias. No entanto, no norte do país representa até 1,83%, semelhante à informação da fisiopatologia dos Estados Unidos 11, 12
HCL é um transtorno crônico linfoproliferativo de células B. No entanto, suas células não têm a aparência de nenhuma sub-população de células B, e sua origem tem sido uma questão de debate., A análise dos genes das regiões variáveis das imunoglobulinas (Ig) é uma ferramenta utilizada para descobrir a origem clonal das células linfóides.13 em mais de 85% dos casos,somos capazes de encontrar mutações somáticas em genes de regiões variáveis Ig de células HCL,14, 15 o que é uma indicação de que as células envolvidas passaram através do centro germinal ou órgãos periféricos linfóides.16 aproximadamente 40% das células HCL Co-expressam múltiplos isotipos de Ig clonalmente relacionados.17
evidência sugere um centro germinal de origem pós-germinal nas células B de memória, devido ao seu perfil de expressão genómica.,18,19 a origem das células B da memória é compatível com a falta de translocação recíproca cromossômica do HCL.20 ausência de CD27 é típico em HCL. Isto representa um ponto contra a hipótese da sua origem nas células B da memória.No entanto, foram observadas células B de memória CD27 negativas.22 Uma vez que o afeto dos gânglios linfáticos é pouco frequente, tem sido sugerido que a célula de origem HCL está provavelmente localizada na medula óssea ou baço, uma vez que esses são geralmente os lugares mais afetados. As células HCL mostram um perfil de expressão semelhante à zona marginal do baço.,23 o aspecto característico de HCL é devido à expressão beta-actina, que é polimerizada Em F-actina, localizada no citoesqueleto cortical.24 PP52 fosfoproteína, que é específica para os leucócitos, está ligada à F-actina e é responsável por trazer apoio às projeções Tipo Cabelo.Por outro lado, a sequenciação do gene HCL identificou recentemente a presença de uma mutação BRAF V600E em quase todos os doentes com a doença, ausente noutras neoplasias linfóides das células B.26,27 mutações BRAF ativam a via MAPK, promovendo crescimento, sobrevivência e diferenciação celular HCL.,A evolução clínica da doença é indolente. A maioria dos pacientes geralmente apresentam fraqueza e fadiga como os sintomas predominantes durante o início da doença.29 por vezes, há uma história de infecções repetidas. Os resultados do exame fisico são: esplenomegalia em 96%, hepatomegalia em 58% e linfadenopatia em apenas 35%. Estes gânglios linfáticos inchados são raramente observados na periferia; no entanto, eles estão geralmente presentes no abdômen e detectados por estudos de imagem.,30
numa fase avançada da doença, somos capazes de encontrar dor no quadrante superior esquerdo, infecções, febre e hemorragias e/ou perda de peso. No entanto, isto é pouco frequente devido à disponibilidade e eficácia do tratamento.As manifestações clínicas são o produto da acumulação de células pilosas no baço, fígado e medula óssea (Tabela 1).32
| Baço | Fígado | medula Óssea | gânglios Linfáticos |
|---|---|---|---|
| As células acumulam-se na polpa vermelha e atrofia a polpa branca. Eles mais tarde formam os chamados “pseudo-sinusóides” através da substituição das células endoteliais da polpa vermelha por canais vasculares melhorados, contribuindo para a anemia. | aqui, acumulam-se nos sinusóides hepáticos, bem como no tracto portal., Há fibrose neste último devido ao abundante ácido hialurônico, que estimula as células cabeludas a produzir fibronectina, com a correspondente fibrose. | nesta área, há extensa produção de fibrose e supressão da hematopoiese. O fator chave é a interação das células pilosas com o ácido hialurônico da matriz extracelular, gerando fatores de crescimento fibroblástico (FGF) e estimulando as células malignas a produzir e segregar fibronectina. | geralmente, ausente da doença. Falta de receptores para a entrada de células cabeludas., |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosis
HCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v)., O último ocorre em 10% dos casos com uma idade média de 70 anos, e apesar das semelhanças com leucemia de células pilosas clássicas, eles divergem na ausência dos marcadores imunofenotípicos CD25 e CD123. Outra forma de fazer um diagnóstico diferencial é a falta de resposta ao tratamento padrão de HCL e a inexistência de mutações do gene BRAF V600F.O diagnóstico de HCL é comumente feito com biópsia e aspiração da medula óssea combinada com caracterização imunofenotípica através da citometria de fluxo.,33 é importante salientar que esta patologia é geralmente sub-diagnosticada e requer suspeita clínica e o uso da tecnologia adequada para resolver este problema. Como mencionado anteriormente, a maioria dos pacientes (70-90%) presente pancitopenia, com leucopenia (
×109/L), anemia (g/dL), neutropenia (×109/L), monocytopenia (×109/L) e trombocitopenia (×109/L). Apenas entre 10% e 20% apresentam leucocitose moderada (
10×109/L). Os doentes com HCL apresentam níveis séricos elevados de IL-2R (CD25), que se correlacionam com o grau de actividade da doença.,Outros testes que devem ser considerados quando se faz o diagnóstico são os níveis de imunoglobulina sérica, bem como o gene IgVH e as mutações somáticas BRAF V600E.32 Alguns HCL histopatológica e immunophenotypic características são as seguintes:
- •
de Linfócitos citometria de fluxo de sangue periférico ou medula óssea com CD19, CD20, FMC7, CD11c, CD103, CD25, HC2, CD22, sIg, CD79a e CD123 expressões, com quatro sendo o principal e marcadores específicos: CD11c, CD103, CD25 e CD123.34 Comumente negativo marcadores são CD5, CD23, CD10, CD79b e CD27.,32
- •
uma forte expressão para CD200 é característica da HCL e pode ser útil no diagnóstico de casos difíceis.A aspiração da medula óssea com uma agulha pode ser difícil de obter e é frequentemente improdutiva ou “seca”. Numa biopsia da medula óssea, podemos observar fibrose, com um olhar de “ovo frito” celular causado pelos amplos espaços entre núcleos e citoplasma abundante. São realizadas análises de imunohistoquímica para CD20 e armadilha (fosfatase ácida resistente ao tartarato), DBA-4 e annein A1, que são caracteristicamente positivas.,32
Andrulis et al. foi realizado um estudo onde foi relatada a eficiência do anticorpo VE1 para detecção de BRAF V600E, juntamente com a identificação de HCL em outras entidades. Além disso, um estudo realizado por Uppal et al. detectou uma sensibilidade de 88% e uma especificidade de 97% para detectar esta mutação com o anticorpo mencionado.O tratamento actual
HCL tem uma evolução indolente. Esperar e observar é uma boa opção para pacientes assintomáticos, uma vez que o tratamento precoce não oferece qualquer tipo de benefício nas taxas de sobrevivência destes casos., De qualquer forma, a progressão da doença na maioria dos pacientes irá levar a complicações como resultado de citopenias e esplenomegalia; ou seja, anemia, hemorragias, infecções recorrentes, etc. Na prática geral, o tratamento deve ser iniciado se ocorrer algum dos critérios listados no quadro 2.Quando for tomada a decisão de não iniciar o tratamento, deve efectuar-se uma monitorização clínica e laboratorial de três em três meses durante o primeiro ano e, posteriormente, de seis em seis meses.,O tratamento com HCL não é considerado curativo, mas as estratégias de tratamento atuais são capazes de alcançar remissões prolongadas, aumentando assim as taxas de sobrevivência global (algoritmo 1).36-39
critérios para iniciar o tratamento.
1. Esplenomegalia sintomática 2. Citopenia envolvendo pelo menos um dos seguintes:
– hemoglobina g/dL
– plaquetas ×109/L
– neutrófilos ×109/L
3., Infecções graves
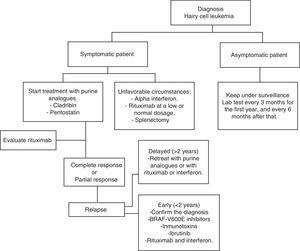
gráfico para o tratamento de um doente com leucemia de células pilosas.
análogos de Purina
Em 1960, os resultados mostraram que 30% das crianças com síndrome de imunodeficiência combinada severa faltou deaminase da adenosina enzima (ADA).,Além disso, descobriram que a acumulação da forma trifosfato de desoxiadenosina foi responsável pela diminuição dos linfócitos.41 tendo estas observações em mente, medicamentos capazes de se ligar irreversivelmente a ADA ou antagonizar a sua acção foram desenvolvidos. Após tratamento com análogos de purina, a acumulação de trifosfato de desoxiadenosina resulta em ruptura e inibição da reparação do ADN, que se traduz em apoptose celular.,42
Pentostatina
também conhecida como 2′ – desoxicoformicina( dcF), um produto de inibidores Streptomyces antibioticus e ADA, introduzido pela primeira vez em 1980 como o primeiro análogo de purina para o tratamento com HCL. Na Tabela 3 37,39,43,44 estudos específicos mostram taxas de resposta à pentostatina. A utilização intravenosa de pentostatina a uma razão de 4 mg/m2, de duas em duas semanas, até se obter uma resposta completa, nos EUA, A Pentostatina é segura em doentes com iluminação da creatinina
60 ml/min. No entanto, é necessária uma redução da dose se esta depuração se situar entre 40 e 60 ml/min.,Recomenda-se hidratação do doente com 1, 5 L de solução intravenosa em cada ciclo de pentostatina.A resposta Global varia entre 88% e 96%, enquanto a resposta completa varia entre 44% e 81%. Flinn et al. estima-se uma taxa de sobrevivência de 5 e 10 anos de 90% e 81%, respectivamente, com uma duração média de seguimento de 9, 3 anos.A Pentostatina é geralmente bem tolerada e os efeitos adversos mais frequentes são anemia, trombocitopenia e neutropenia.,Ainda assim, foi relatado que a pentostatina diminui significativamente a contagem de linfócitos CD4+ e CD8+, o que pode aumentar as neoplasias secundárias e a incidência de infecções.47,48 cladribina
é conhecida como 2-clorodeoxiadenosina (CdA). Na tabela 4,49-52 os estudos indicam sua eficácia. O esquema mais utilizado consiste na administração de 0, 1 mg/kg/dia em perfusão contínua durante 7 dias. Num estudo não aleatorizado, houve evidências que provaram que não houve diferença estatisticamente significativa nos intervalos de resposta e toxicidade entre as perfusões (24h e 2h).,Outro estudo aleatorizado comparou a administração diária versus semanal de cladribina; não houve resultados significativos nas taxas de resposta, sobrevivência, global e toxicidade.Um estudo diferente demonstrou que o programa semanal reduziu o risco de infecções.Uma das vantagens da administração subcutânea é o facto de, na maioria dos casos, não necessitar de hospitalização. 0,14 mg/dia durante 5 dias tem uma taxa de resposta de 95%, 56 semelhante à administração intravenosa. Os programas subcutâneos semanais têm taxas de resposta e toxicidade semelhantes às diárias.,57 com apenas um ciclo de cladribina, uma resposta global até 100% pode ser obtida, e as taxas de resposta total diferem de 77% para 95%.49-52 Jehn et al. relatou uma sobrevivência global aos 12 anos de 79%.Em geral, a cladribina é bem tolerada, sendo as citopenias e a febre os efeitos adversos mais frequentes.
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days |
77.3 | 18.,6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days |
88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days |
95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.,1mg/kg/d 7 days |
79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence., Mesmo assim, existem estudos retrospectivos que provam que ambos os medicamentos têm uma eficácia semelhante, em termos de resposta completa e sobrevivência livre de doença.outros tratamentos análogos da purina continuam a ser a primeira linha de tratamento, mas novas descobertas sobre a patofisiologia da HCL levaram à criação de fármacos com diferentes alvos terapêuticos. Estes medicamentos estão sob investigação e alguns revelaram resultados promissores.
Rituximab
Uma vez que o HCL é uma doença maligna das células B, é lógico utilizar um anticorpo monoclonal contra o CD20, tal como o rituximab., Utilizado como fármaco isolado, o rituximab pode atingir taxas de resposta total de 10-54% em doentes com recidiva de HCL, numa dose de 375 mg/m2, uma vez por semana, durante 4-8 semanas.58,59 Else et al. reviu retrospectivamente 18 doentes que foram tratados com análogos da purina em associação com rituximab como segunda linha de tratamento, após serem tratados com análogos da purina como agentes únicos. Todos os doentes responderam, com uma taxa de resposta completa de 89%.60
Rituximab a 375 mg / m2 por semana durante 8 semanas como terapêutica inicial após administração de 5.,6 mg/m2 de cladribina por perfusão intravenosa de 2-h durante 5 dias gera uma resposta total de 100%.Em situações especiais ou desfavoráveis, podem ser utilizados 100 mg de rituximab por semana durante 4 a 6 semanas. Este facto é menos dispendioso e é geralmente eficaz, especialmente quando associado ao interferão.embora os análogos da purina possam não ser capazes de eliminar a HCL, uma vez que a doença residual mínima detectada após a administração de cladribina é sempre fortemente CD20+, a erradicação da MRD pode ser obtida utilizando rituximab. Rivandi et al., demonstrou, num estudo preliminar, que o rituximab, nas doses convencionais durante um período de 8 semanas, actua com grande actividade, eliminando a DRM em 13 doentes, quando é utilizado 4 semanas após a administração de cladribina.Como descrito anteriormente, a mutação BRAF V600E é a chave genética na HCL. Portanto, é um alvo terapêutico que foi estudado nos últimos anos. O Vemurafenib é um inibidor oral da BRAF V600E. Tiacci et al., conduziu um estudo para medir a actividade e segurança do Vemurafenib em doentes com CPPC que recidivaram após tratamento com análogos da purina ou que se refractaram à administração de análogos da purina. A taxa de resposta Global foi de 96% e a taxa de resposta completa foi de 35%, com uma média de sobrevivência sem recaídas de 19 meses. Os efeitos adversos foram erupção cutânea, artralgia e artrite.Uma vez que foi observada uma relação positiva entre a utilização de Vemurafenib e o aparecimento de doenças malignas dermatológicas, são recomendadas explorações frequentes da pele.,Ibrutinib um inibidor selectivo e irreversível da tirosina cinase de Burton intervém na Via sinalizadora das células B.Iniciou-se recentemente um ensaio clínico do ibrutinib em doentes com recidiva de HCL. Os dados preliminares de eficácia e segurança mostram efeitos adversos tais como erupções, diarreia e artralgia. Este ensaio clínico está atualmente ocorrendo em vários centros nos EUA (NCT01841723).Imunotoxinas
de modo a aumentar a citotoxicidade monoclonal dos anticorpos, foram criadas técnicas que facilitam a produção de conjugados de anticorpos–toxina ou anticorpos–fármacos., Uma imunotoxina é a fusão entre uma toxina bacteriana (isto é, Pseudomonas exotoxina ou difteria) e a fracção variável de um anticorpo monoclonal, cujo alvo específico se encontra na superfície de células neoplásicas como CD25 ou CD22. Esta toxina é libertada no interior da célula neoplásica e interfere com a síntese proteica.65
BL22 é uma imunotoxina contra o CD22 fundida com uma forma truncada da P. exotoxina PE38. Num ensaio clínico de fase II, O BL22 foi testado em 36 casos de recidiva de HCL ou de doença refractária., Após um ciclo (40 mg/kg de dois em dois dias, três doses), a taxa de resposta completa foi de 25% e a taxa de resposta global foi de 50%. Estas respostas melhoraram para uma taxa de resposta total de 47% e uma taxa de resposta global de 72% após o retratamento (apenas em doentes com citopenias). Dois doentes desenvolveram síndrome hemolítica uremica sem necessidade de recorrer à plasmaférese.Subsequentemente, o moxetumomab pasudotox foi desenvolvido como uma versão modificada do BL22 com uma maior afinidade e citotoxicidade., Num ensaio de fase I, que incluiu 28 doentes com recidiva e resistência ao HCL, Obteve-se uma taxa de resposta global de 86%, incluindo uma taxa de resposta completa mantida em 46% dos doentes.67 opções terapêuticas em circunstâncias desfavoráveis embora a HCL seja tratada na maioria dos países desenvolvidos com cladribina e pentostatina, é um facto que estas drogas não são apenas caras, não estão disponíveis no México e em muitos países com recursos limitados., Assim, nestes tipos de circunstâncias, existem outras opções terapêuticas acessíveis com resultados favoráveis ao tratamento com interferão alfa para o tratamento de doentes com HCL foi introduzido pela primeira vez em 1984. Hoje, seu uso é limitado, principalmente devido à grande eficácia dos análogos de purina. Por outro lado, nos países com baixos recursos económicos, é uma opção barata e que provou resultados semelhantes aos da cladribina em termos de sobrevivência global. Ruiz-Delgado et al., realizou um estudo comparativo entre o interferão-alfa (n=18) e cladribina (n=11), onde a diferença na sobrevivência global entre os dois grupos não foi estatisticamente significativa; 94% em 217 meses em que o interferon grupo e 91% a 133 meses no cladribina grupo.68 em um estudo realizado em nosso centro, nove pacientes com HCL receberam três mega-unidades IFN três vezes por semana durante 12 semanas, posteriormente eles receberam tratamento mais uma vez durante 8 semanas quando houve reativação de leucemia ou após 10 meses de observação a cada ano., Todos os doentes tinham uma remissão hematológica antes das 12 semanas de tratamento. Esta opção terapêutica é mais barata, eficaz e comparável a outras formas de terapia com IFN no tratamento e manutenção de pacientes com este tipo de leucemia.É possível combinar o interferão com rituximab sem aumentar os efeitos tóxicos e melhorar a eficácia.a esplenectomia foi a primeira intervenção que alterou significativamente a sobrevivência dos doentes. Hoje, é raramente usado., Pode ser recomendado em doentes com esplenomegalia maciça dolorosa (>10cm sob o bordo costal) e com infiltração mínima da medula óssea, ou em doentes refractários ao tratamento com análogos do interferão e purina.33 estudos retrospectivos mostram uma taxa de resposta completa de 40-62%, e uma taxa média de sobrevivência de 5 anos até 68%.70,71 Lad et al. publicou um estudo retrospectivo, incluindo 24 doentes com diagnóstico de HCL, que foram divididos em dois grupos: 17 doentes receberam cladribina e 7 foram esplenectomizados., 75% dos pacientes no grupo de esplenectomia apresentaram remissão total, 94% no grupo de cladribina. Um achado interessante ao comparar ambos os grupos foi que não foram observadas diferenças estatisticamente significativas em relação à sobrevivência livre de leucemia e sobrevivência global.72 prognóstico
O tempo de sobrevivência em doentes após o diagnóstico foi de 4 anos antes de se conhecer um tratamento, devido a complicações derivadas de citopenias, incluindo hemorragias e infecções. Posteriormente, com esplenectomia como tratamento de primeira linha, houve uma resposta completa de 40-62% e uma taxa de sobrevivência de 5 anos com 61-68%., Em seguida, o interferão alfa foi utilizado como a primeira droga com benefícios no tratamento de HCL. Ainda assim, a sua taxa de resposta total foi baixa, com 10%.Actualmente, com análogos da purina (pentostatina e cladribina), é induzida uma resposta completa até 80% dos doentes com uma sobrevivência mediana de 10 anos. As taxas de resposta Global são de 96-100% com uma taxa de resposta completa de 80% e uma taxa de sobrevivência de 10 anos variando entre 85% e 100%.Apesar disso, uma proporção significativa de doentes com HCL falha na resposta ao tratamento ou torna-se resistente., Até 48% dos doentes recidivaram nos 15 anos seguintes.O futuro dos doentes com HCL é muito favorável. O desafio é identificar esta malignidade o mais cedo possível e tratá-la adequadamente usando os recursos disponíveis.conflito de interesses
os autores não têm conflitos de interesses a declarar.