i föregående inlägg introducerade vi ortho- ,para – Och meta – directors i elektrofil aromatisk substitution. Tidigare till att vi täckte mekanismen för elektrofil aromatisk substitution, och visade att mekanismen fortskrider genom en karbokation mellanliggande.,
idag kommer vi att knyta dessa två begrepp tillsammans, och försöka visa att huruvida en substituent är en ortho -, para – eller meta – regissör beror på hur substituenten påverkar stabiliteten hos karboceringsmediären.
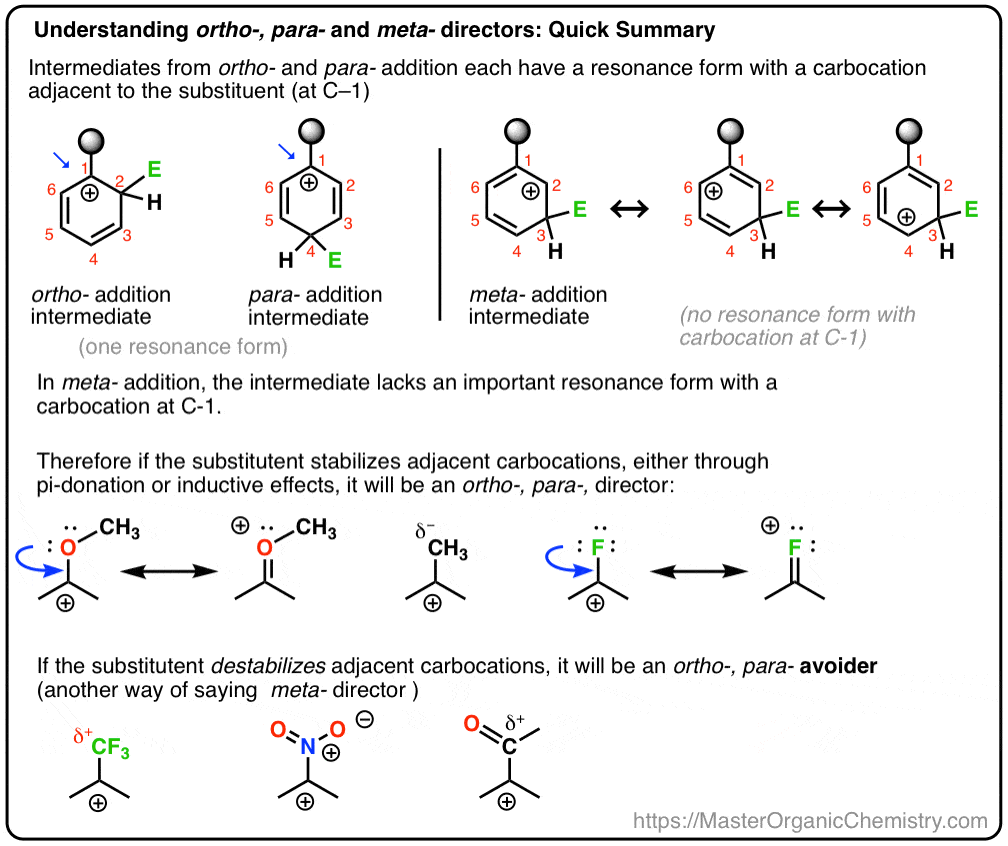
och vi kommer också att se varför ett alternativt namn för meta – director kan vara ”ortho -, para – avoider”. :- )
Innehållsförteckning
- karbokationer stabiliseras av intilliggande atomer med Lone Pairs
- fallstudie # 1: An ortho-,para – Director (och3)
- ja, syre är elektronegativ., Men Donation Av Sitt Ensamma Par Mer Än Gör Upp För Det!
- reaktions-Energidiagrammet för en Ortho Para – direktör
- fallstudie #2: en meta-direktör (CF3).
- vad vi kallar ”meta– Directors” är mer som ”ortho -, para-Avoiders”
- Reaktionsenergidiagrammet för en meta-Director
- så varför är Halogener Ortho -, Para-Directors?
- anteckningar
- Quiz själv!,
- (avancerade) referenser och ytterligare läsning
karbokationer stabiliseras av intilliggande atomer med ensamstående par
låt oss tänka tillbaka till vilka faktorer som ökar stabiliteten hos karbokationer.
karbokationer är elektronfattiga arter med sex elektroner i sitt valensskal. Så, någon substituent som kan donera elektrondensitet mot karbocen kommer att stabiliseras. Detta inkluderar:
- elektronfrisättande substituenter som kan fungera som ”sigma-donatorer” (t. ex., många alkylgrupper, som donerar elektrondensitet genom induktiva effekter)
- intilliggande atomer med ensamma par som kan fungera som ”pi–givare” till karbokatet
- intilliggande c-c pi-bindningar som kan stabilisera karbokatet genom laddningsavlokalisering (”resonansstabilisering”, med andra ord).
och faktorer som destabiliserar carbocations?
- Alla substituenter som avlägsnar elektrondensitet från en karbocation, såsom starka elektronutdragande grupper.
Så här är en snabb frågesport., Med tanke på allt skrivet ovan, vilken av de två karbokaten nedan kommer att vara stabilare?
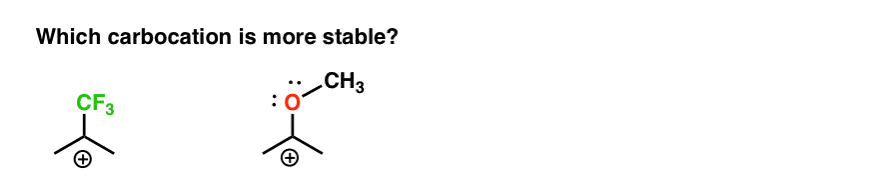
om du kan svara på den här frågan Är du mest på väg mot att förstå skillnaden mellan ortho -, para – och meta – regissörer.
låt oss undersöka två fallstudier. Vi tittar på en generisk elektrofil aromatisk substitutionsreaktion av bensen med en Orto-, para – regissör (metoxibensen) och undersöker de intermediärer som erhålls från attack på Orto -, meta-och para-positionerna., Då ska vi utföra samma övning med en meta-regissör (trifluorometylbensen).
i processen hoppas vi förstå varför metoxibensen gynnar Orto – och para– produkter medan trifluorometylbensen gynnar metaprodukten.
fallstudie #1: an ortho-,para – Director (och3)
elektrofil aromatisk substitution av metoxibensen tenderar att ge ortho – och para – produkter med mycket lite meta- ., Här är vad som händer i nitrering, till exempel:
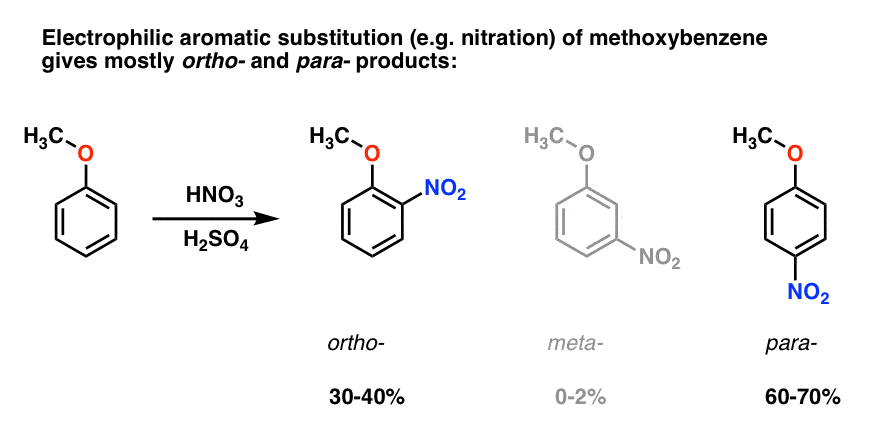
para – produkten dominerar (60-70%), med ortho – nära bakom (30-40%) och bara ett spår av meta– .
varför?
3. Ja, Syre Är Elektronegativ. Men Donation Av Sitt Ensamma Par Mer Än Gör Upp För Det!
här är vägen som leder till ortho – produkten., Vi bryter en c-c pi-bindning och bildar en ny C–E-bindning (där E är avsedd att representera en generisk elektrofil) och genererar en resonansstabiliserad karbocation:
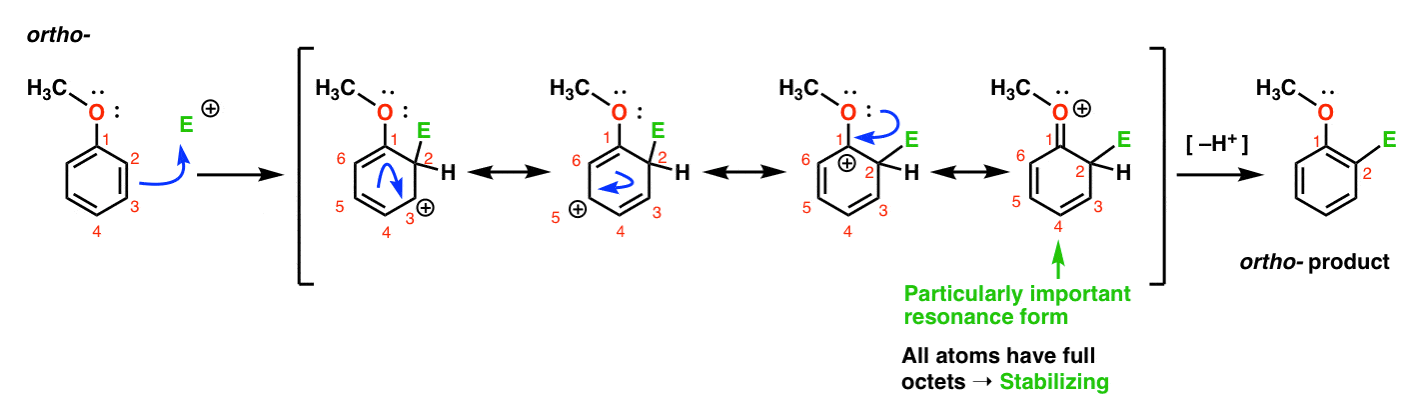
om du tittar noga bör du notera att vi kan bilda fyra resonansformer . Tre av dem bär fulla karbokationer (på C1, C3 och C5), men det är den fjärde – resonansformen där det finns en c=o pi – bindning-vilket är särskilt viktigt. Varför? I denna resonansform har alla kolatomer en full oktett av elektroner., Det beror på att syret direkt bundna till ringen kan donera en ensam par till den intilliggande carbocation, bildar en pi bond. Detta är ett exempel på pi donation.
här är vägen för elektrofilens attack vid metapositionen. Notera skillnaden!
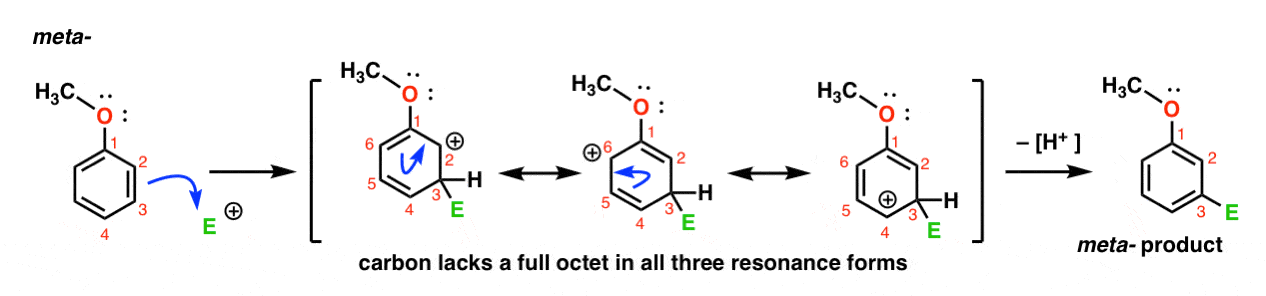
elektrofilens Attack vid C-3 resulterar i en karbocation som kan delokaliseras via resonans till C2, C4 och C6. Det finns dock inget sätt att ”flytta” karbokatet till C1, vilket innebär att det inte finns någon rimlig resonansform där alla atomer har fulla oktetter.,
detta gör meta – carbocation intermediate mycket mindre stabil än ortho – carbocation intermediate.
slutligen, här är reaktionen som leder till para – produkten:
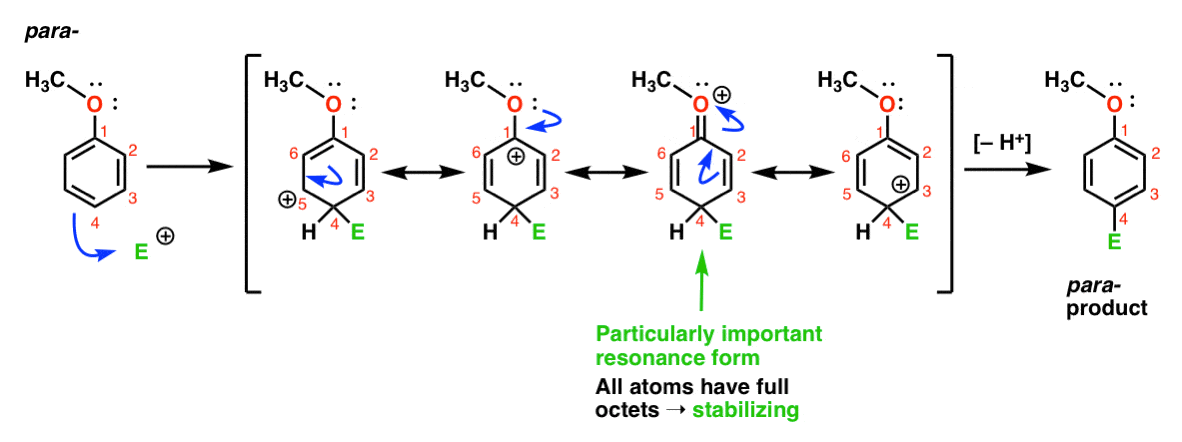
Här kan vi återigen rita resonansformer med karbokationer på C1, C3 och C5, samt en fjärde resonansform där den bifogade syreatomen donerar ett elektronpar till förgasningen på C1, vilket resulterar i en full oktett vid kol. Detta är en situation som i huvudsak är densamma som ortho – mellanliggande.,
så, genom analys av resonansformerna (och markera mitt ord, kan du väl bli ombedd att göra detsamma på ett test inom en snar framtid) kan vi se att de mellanliggande karbokationerna som härrör från ortho – och para– addition är betydligt stabilare än mellanliggande från meta– addition.
detta förklarar varför ortho – och para – produkter dominerar, och meta – produkten är mindre.
det kan vara bra att visualisera detta genom att rita ett reaktionsenergidiagram.
4., Reaktions-Energidiagrammet för en Ortho Para-direktör
övergångstillståndet som leder till ortho – och para-additionsprodukterna är mycket lägre i energi än meta -.
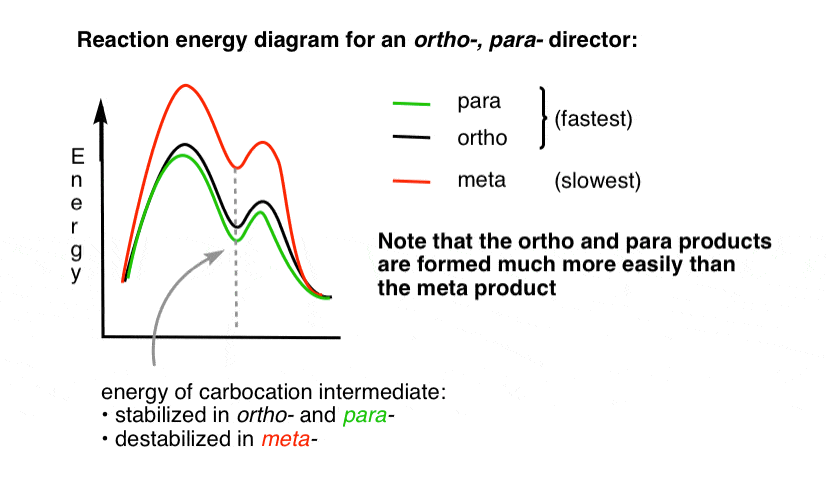
en fråga – Varför tror du att para-kan gynnas (t. ex. 60% -70% utbyte för para– produkten av nitrering, ovan, jämfört med 30-40% för ortho -) även om det finns två ortho – positioner och bara en para -?
Tänk på det i en minut, och vi kommer att ta itu med det nedan.
5. Fallstudie # 2: en metaregissör (CF3).
Så vad sägs om CF3?, Varför ger det meta-produkter? Låt oss göra samma typ av analys.
här är vägen som leder till ortho – produkten. Som med ortho-mellanliggande ovan kan vi rita tre resonansformer som placerar karbokatet på C1, C3 respektive C5.
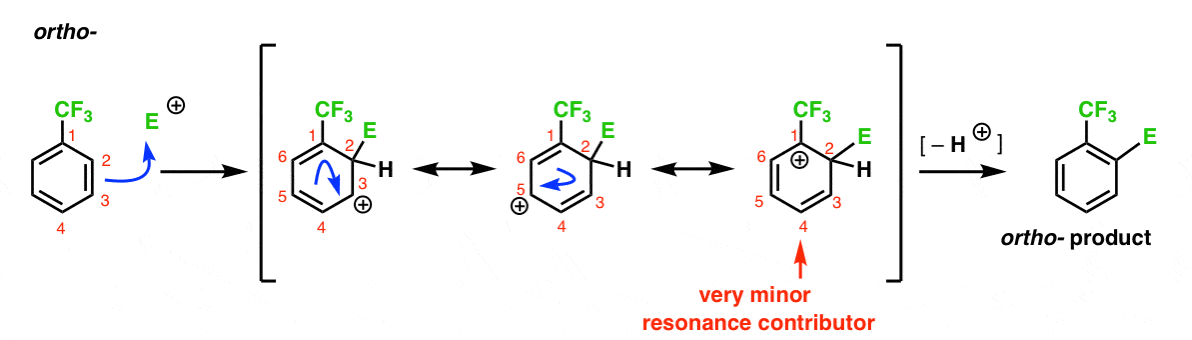
Så vad är annorlunda i det här fallet?
det som är annorlunda är närvaron av en stark elektronutdragande grupp (CF3) vid C1, och detta förändrar helt bollspelet.
som vi har sagt destabiliseras carbocations av grannar som drar tillbaka elektrondensitet., Därför förväntar vi oss att detta är en mycket mindre resonans bidragsgivare, med resultatet att den positiva laddningen endast delokaliseras över C3 och C5.
Vi har noterat att CF3 är en metaregissör. Det är rimligt att tro att det finns något om meta – som gör det särskilt stabilt. Genom att analysera intermediären för metaprodukten nedan kan du se varför?
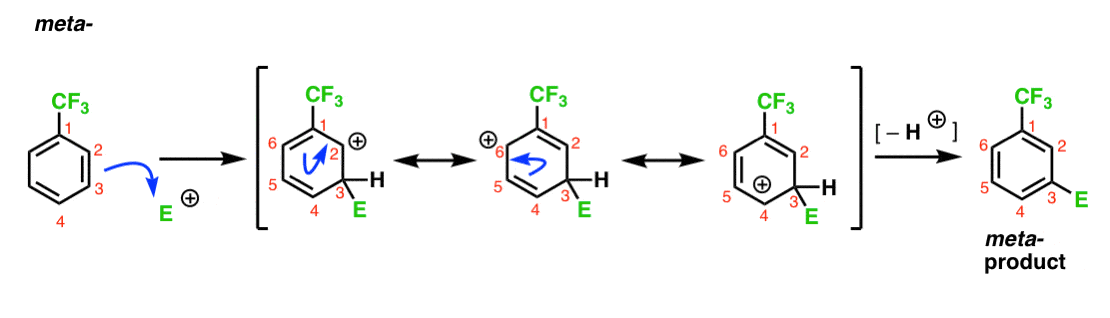
hmmm. Det finns ingen intermediär där ett kol har en full oktett. Det finns inga särskilt stabila resonansformer.
det är typ av ”meh”, eller hur?
6., Vad vi kallar ”meta– Directors” är mer som ”ortho -, para-Avoiders”
å andra sidan finns det inte heller några särskilt instabila resonansformer. Eftersom den positiva laddningen är lokaliserad på C2, C4 och C6 undviks en situation där den positiva laddningen ligger direkt intill elektronutdragningsgruppen CF3. Och den positiva laddningen är sålunda delokaliserad snyggt genom hela ringen, till skillnad från i ortho – situationen ovan.
så det är en ”mindre dålig” mellanliggande än ortho-.,
slutligen har para-intermediären samma problem som ortho- ; en mellanliggande med en positiv laddning på C1, intill CF3.
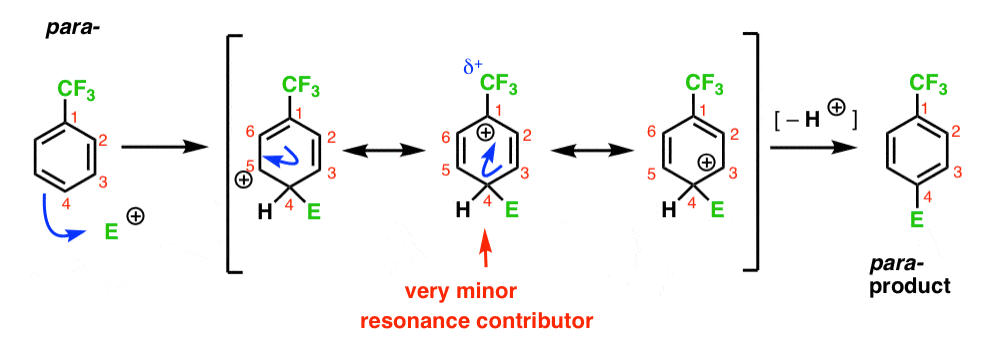
detta resulterar i endast två viktiga resonansformer, vilket leder till en mindre delokaliserad (och därför mindre stabil) förgasningsmediär.
nedre raden: meta– karboceringsmediären är stabilare än antingen ortho – eller para-intermediärerna.
men det beror inte på att substituenten själv har någon stabiliserande effekt på meta– mellanliggande. .,
Jag skulle hävda att det är mer användbart att tänka på meta – som mindre instabil än ortho – och para– .
På detta sätt är det inte så mycket att CF3 är en metaregissör, så mycket att det är en ”ortho -, para– avoider. ”
7. Reaktionsenergidiagrammet för en metaregissör
här är en skiss av reaktionsenergidiagrammet:
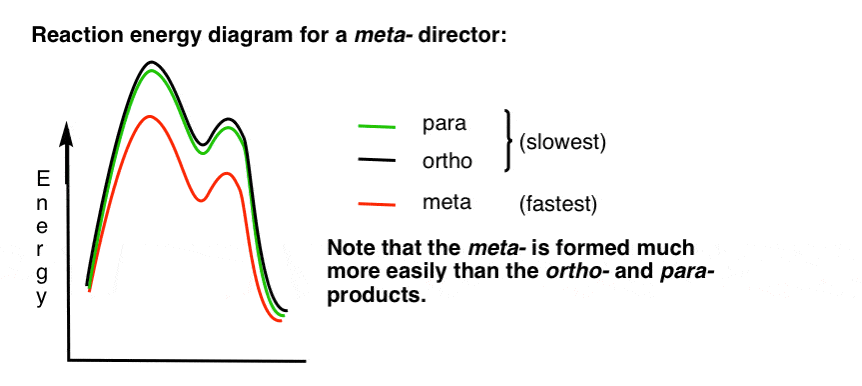
okej.
tillbaka till vår ursprungliga fråga. Så vilken karbokation är stabilare?,
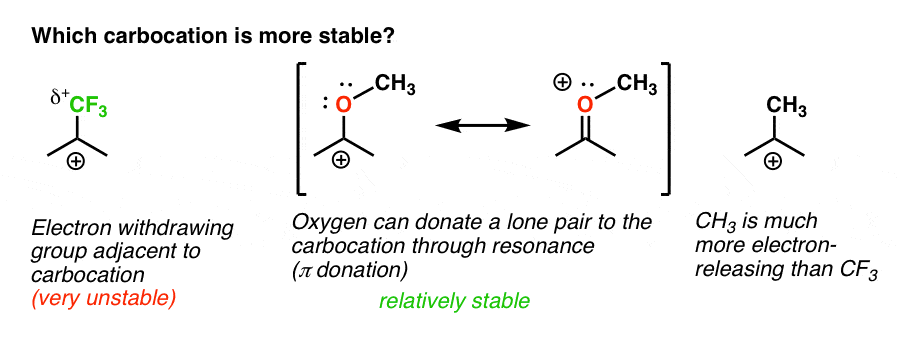
det är tydligt att förgasningen med ett intilliggande syrelager med ett ensampar är mycket stabilare än en förgasning intill en elektronutdragande grupp som CF3.
och det innefattar skillnaden mellan en ortho-, para – director som och3 och en meta – director som CF3.
det är värt att notera att de flesta alkylgrupper (som CH3 ) saknar ett ensam par, men är fortfarande ortho-, para – directors., Detta överensstämmer med allt vi har lärt oss tidigare om hur alkylgrupper i allmänhet stabiliserar för karbokationer-minns att karbokationsstabiliteten generellt ökar med substitution
Så om dessa para– produkter…
varför tillverkas para – produkter i högre takt än ortho- ?
svaret är steriskt hinder.
Attack vid para – positionen är mindre belastad av substituenten än attack vid ortho-positionerna är.
så varför är Halogener Ortho -, Para-Directors?
detta lämnar oss med det något knepiga exemplet på halogener.,
Varför är fluor, klor, brom och jod Orto -, para – direktörer trots att de inaktiverar grupper?
svaret bör komma som ingen överraskning om du har följt med, men vi kommer att lämna detta till nästa inlägg, eftersom det här är tillräckligt länge redan.
Nästa inlägg: Varför är halogener ortho -, para-direktörer?
anmärkningar
anmärkning 1. Det är mer korrekt att säga att ortho – och para-produkter dominerar eftersom övergångsstaterna som leder till dessa produkter är lägre i energi, snarare än intermediärernas energier själva., Det är trots allt energin i övergångsstaterna som bestämmer aktiveringsbarriären, och därför reaktionshastigheten.
Anmärkning 2. Eller hyperkonjugation, som de flesta läroböcker (med det anmärkningsvärda undantaget Maitland Jones) i allmänhet undviker.
Quiz själv!,/div>
 Klicka för att Knäppa
Klicka för att Knäppa
Klicka för att Knäppa  Klicka för att Knäppa
Klicka för att Knäppa 
(Avancerad) Referenser och lästips
- A., F. Holleman, Dö direkte Einführung von Substituenten i den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/recl.19100291205
A. F Holleman från 1910 sade att ortho–para läggning i samband med aktivering och meta läggning med släckningen. - – arten av den alternerande effekten i kolkedjor. Del XXIII. avvikande orientering av halogener, och dess inverkan på problemet med Ortho–para-förhållandet, i aromatisk substitution
Christopher Kelk Ingold och Charles Cyril Norrey vass
J. Chem. Soc. 1928, 417-425
DOI: 10.,1039 / JR9280000417
i detta dokument diskuteras riktningseffekter i 1,2-dihalobensener. - några observationer rörande steriska hinder och effekterna av substituenter på Ortho : para-förhållandet i aromatisk substitution
P. B. D. De la Mare
J. Chem. Soc. 1949, 2871-2874
DOI: 10.1039/JR9490002871
I denna uppsats, de la Mare diskuterar olika faktorer som kan stå för en dominans av para över ortho selektivitet, efter att dividera avkastningen av ortho produkten av 2 (eftersom det är 2 ortho positioner men bara 1 para position i monosubstituted bensener)., - Volymeffekter av alkylgrupper i aromatiska föreningar. Del V monosulfonering av p-cymene
R. J. W. Le Fèvre
J. Chem. Soc. 1934, 1501-1502
DOI: 10.1039 / JR9340001501
i P-cymen är huvudprodukten som erhålls vid elektrofil sulfonation 2-produkten (Orto till metylgruppen), sannolikt på grund av sterik. - effekter av alkylgrupper i elektrofila tillsatser och substitutioner
COHN, H., HUGHES, E., JONES, M. och PEELING, M. G.
Nature 1952, 169, 291
DOI: 1038 / 169291a0
detta dokument har data som jämför nitreringen av T-butylbensen och toluen., T-butylbensen är mycket mer p-riktad än toluen (79.5% para För t-butylbensen vs. 40% para för toluen), vilket sannolikt beror på sterik (ortho-tillvägagångssättet blockeras av bulkier t-butylgruppen). - Distribution av isomerer vid Mononitrering av etyl – och Isopropylbensen. Ytterligare ett Bevis för en Sterisk Effekt i Isomer Distribution
Herbert C. Brown och W. Hallam Bonner
Journal of American Chemical Society 1954, 76 (2), 605-606
DOI: 10.,1021 / ja01631a084
tabell II i detta dokument visar att ortho-produkten som erhålls genom nitrering av monoalkylbensener minskar när alkylgruppen blir större (t.ex. ger t-butylbensen mycket liten Orto-produkt vid nitrering jämfört med toluen). - – arten av den alternerande effekten i kolkedjor. Del XXII. ett försök att ytterligare definiera den sannolika orienteringsmekanismen i aromatisk substitution
Christopher Kelk Ingold och Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926
DOI: 10.,1039 / JR9270002918
ett tidigt papper av den inflytelserika fysiska organiska kemisten, Prof.C. K. Ingold, som anger att halogenobensener induktivt elektronutdragande men samtidigt resonansstabiliserande. - den avvikande reaktiviteten hos Fluorobensen i elektrofil aromatisk Substitution och relaterade fenomen
Joel Rosenthal och David I., Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 1021 / ed080p679
ett mycket intressant papper, Lämplig för nyfikna undergrads, och diskuterar något som de flesta praktiserande organiska kemister kommer att veta empiriskt-fluorobensen är nästan lika reaktiv som bensen i EAS eller Friedel-hantverk reaktioner, vilket är kontraintuitivt när man överväger elektroniska effekter! - organisk kemi
N. Haworth, C. K. Ingold, T. A. Henry
Ann. Rep. Prog. Chem. 1926, 23, 74-185
DOI: 10.1039 / AR9262300074
ett tidigt dokument som diskuterar rikta effekter i EAS. PP., 136 och 137 innehåller siffror som visar flödet av elektroner. Pg. 140 diskuterar meta-riktning, som uppstår av ”motsatta” skäl som o / p-riktning av alkylgrupper.