Introducción
la leucemia de células pilosas (HCL) es una neoplasia linfoide de células B. El HCL se diferencia de otras neoplasias de células B por sus características morfológicas, inmunofenotípicas y moleculares. Su principal característica es la acumulación de células B monoclonales, con proyecciones en forma de pelo en su superficie, que se encuentran principalmente en la sangre periférica, la médula ósea y el bazo. Fue descrito en 1958 por Bournoncle et al. Sin embargo, lo llamaron reticuloendoteliosis leucémica.,1 El término «células pilosas» fue acuñado en 1996 por Schrek et al.2 hoy en día está clasificado por la Organización Mundial de la salud como un linfoma No Hodgkin de células B.3
definición
el HCL es un trastorno linfoproliferativo crónico poco frecuente de células B. Sus principales características son las prolongaciones citoplasmáticas que dan a las células un aspecto peludo.
etiología
aunque existen diferentes estudios que muestran una relación entre la exposición a ciertos agentes y el HCL, su etiología no ha sido claramente establecida, lo que podría explicarse por su baja incidencia., Entre los agentes que han demostrado una relación positiva están: exposición a pesticidas, 4 herbicidas, aceites minerales, trabajo como carpintero o agricultor.4-7 recientemente se ha descrito una relación positiva con el tamaño5,mientras que el tabaquismo tiene una asociación inversa, especialmente en hombres.8 se desconoce el mecanismo específico que confiere la protección; sin embargo, se ha sugerido que el tabaquismo disminuye la gravedad de los mecanismos inflamatorios9 y que la nicotina induce la apoptosis en los linfocitos.10
Epidemiología
el HCL representa 2-3% de todas las leucemias. Hay 600 nuevos casos reportados por año en los Estados Unidos (3.,2 casos por millón de habitantes). La mediana de edad en el momento del diagnóstico es de 52 años y es más común en hombres que en mujeres, con una proporción de 4:1; con una mayor incidencia en la población blanca, especialmente entre los judíos Ashkenazi.3 en México, el HCL representa el 1,12% de todas las leucemias. Sin embargo, en el norte del país representa hasta el 1,83%, similar a la información de los EE.UU. 11, 12
Fisiopatología
el HCL es un trastorno crónico linfoproliferativo de células B. Sin embargo, sus células no tienen la apariencia de ninguna subpoblación de células B, y su origen ha sido un tema de debate., El análisis de genes de regiones variables de inmunoglobulinas (Ig) es una herramienta utilizada para descubrir el origen clonal de las células linfoides.13 en más del 85% de los casos, podemos encontrar mutaciones somáticas en genes de regiones variables Ig de células HCL,14,15, lo que indica que las células involucradas han pasado por el centro germinal o por órganos periféricos linfoides.16 aproximadamente el 40% de las células HCL coexpresan múltiples isotipos de Ig relacionados clonalmente.17
La evidencia sugiere un origen post germinal en las células B de memoria, debido a su perfil de expresión genómica.,18,19 el origen de las células B de memoria es compatible con la falta de translocación cromosómica recíproca del HCL.20 la ausencia de CD27 es típica en el HCL. Esto representa un punto contra la hipótesis de su origen en las células B de memoria.21 sin embargo, se han observado células B de memoria CD27 negativas.22 dado que la afección de los ganglios linfáticos es infrecuente, se ha sugerido que la célula de origen del HCL probablemente se encuentre en la médula ósea o el bazo, ya que estos suelen ser los lugares más afectados. Las células HCL muestran un perfil de expresión similar a la zona marginal del bazo.,23
el aspecto característico del HCL se debe a la expresión de beta-actina, que se polimeriza a f-actina, ubicada en el citoesqueleto cortical.24 PP52 la fosfoproteína, que es específica para los leucocitos, está vinculada a la f-actina y es responsable de dar soporte a las proyecciones similares al pelo.25 por otro lado, la secuenciación del gen HCL recientemente identificó la presencia de una mutación BRAF V600E en casi todos los pacientes con la enfermedad, ausente en otras neoplasias linfoides de células B.26,27 mutaciones en BRAF activan la vía MAPK, promoviendo el crecimiento, la supervivencia y la diferenciación de células HCL.,28
presentación clínica y hallazgos de laboratorio
la evolución clínica de la enfermedad es indolente. La mayoría de los pacientes suelen presentar debilidad y fatiga como síntomas predominantes durante el inicio de la enfermedad.29 A veces, hay un historial de infecciones repetidas. Los hallazgos del examen físico son: esplenomegalia en 96%, hepatomegalia en 58% y linfadenopatía en solo 35%. Estos ganglios linfáticos inflamados rara vez se observan en la periferia; sin embargo, generalmente están presentes en el abdomen y se detectan mediante estudios de imagen.,30
en una etapa avanzada de la enfermedad podemos encontrar dolor en el cuadrante superior izquierdo, infecciones, fiebre, hemorragias y/o pérdida de peso. Sin embargo, esto es poco común debido a la disponibilidad y eficacia del tratamiento.30,31 las manifestaciones clínicas son producto de la acumulación de células pilosas en el bazo, el hígado y la médula ósea (Tabla 1).32
las manifestaciones Clínicas.,
| Bazo | Hígado | médula Ósea | ganglios Linfáticos |
|---|---|---|---|
| Las células se acumulan en la pulpa roja y la atrofia de la pulpa blanca. Más tarde forman los llamados «pseudo-sinusoides» a través del reemplazo de las células endoteliales de la pulpa roja por canales vasculares mejorados, contribuyendo a la anemia. | aquí, se acumulan en los sinusoides hepáticos, así como en el tracto portal., Hay fibrosis en este último debido a la abundante ácido hialurónico, que estimula a las células pilosas a producir fibronectina, con su correspondiente fibrosis. | en esta área, hay una amplia producción de fibrosis y supresión de la hematopoyesis. El factor clave es la interacción de las células pilosas con el ácido hialurónico de la matriz extracelular, generando factores de crecimiento fibroblásticos (FGF) y estimulando a las células malignas a producir y secretar fibronectina. | generalmente, ausente de la enfermedad. Falta de receptores para la entrada de células pilosas., |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosis
HCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v)., El último ocurre en el 10% de los casos con una mediana de edad de 70 años, y a pesar de las similitudes con la leucemia de células pilosas clásica, divergen en ausencia de los marcadores inmunofenotípicos CD25 y CD123. Otra forma de hacer un diagnóstico diferencial es la falta de respuesta al tratamiento estándar de HCL y la inexistencia de mutaciones del gen BRAF V600F.
métodos de diagnóstico
el diagnóstico de HCL se realiza comúnmente con una biopsia y aspiración de médula ósea combinada con caracterización inmunofenotípica a través de citometría de flujo.,33 es importante señalar que esta patología suele ser sub-diagnosticada y requiere sospecha clínica y el uso de la tecnología adecuada para resolver este problema. Como se mencionó anteriormente, la mayoría de los pacientes (70-90%) presentan pancitopenia, con leucopenia (
×109/L), anemia (g/dL), neutropenia (×109/L), monocytopenia (×109/L) y trombocitopenia (×109/L). Solo entre el 10% y el 20% presentan leucocitosis moderada (>10×109/L). Los pacientes con HCL muestran niveles séricos elevados de IL-2R (CD25), lo que se correlaciona con el grado de actividad de la enfermedad.,34 otras pruebas que deben considerarse al hacer el diagnóstico son los niveles de inmunoglobulina sérica, así como las mutaciones somáticas del gen IgVH y BRAF V600E.32 algunas características histopatológicas e inmunofenotípicas del HCL son las siguientes: citometría de flujo de linfocitos
- •
en sangre periférica o médula ósea con expresiones CD19, CD20, FMC7, CD11c, CD103, CD25, HC2, CD22, Sig, CD79a y CD123, siendo cuatro los marcadores principales y específicos: CD11c, CD103, CD25 y CD12334.CD23, CD10, cd79b y CD27.,32
i • *
una fuerte expresión de CD200 es característica del HCL y puede ser útil en el diagnóstico de casos difíciles.34
i • *
La aspiración de médula ósea con una aguja puede ser difícil de obtener y con frecuencia es improductiva o»seca». En una biopsia de médula ósea, podemos observar fibrosis, con un aspecto celular de «huevo frito» causado por los amplios espacios entre los núcleos y el citoplasma abundante. Se realizan análisis inmunohistoquímicos para CD20 y TRAP (fosfatasa ácida resistente al tartrato), DBA-4 y anexina A1, que son característicamente positivos.,32
Andrulis et al. dirigió un estudio donde se reportó la eficiencia del anticuerpo VE1 para la detección de BRAF V600E, junto con la identificación de HCL en otras entidades. Además, un estudio realizado por Uppal et al. se encontró una sensibilidad del 88% y una especificidad del 97% para detectar esta mutación con el anticuerpo mencionado.34
El tratamiento actual
HCL regularmente tiene una evolución lenta. Esperar y observar es una buena opción para los pacientes asintomáticos, ya que el tratamiento temprano no ofrece ningún tipo de beneficio en las tasas de supervivencia de estos casos., De todos modos, la progresión de la enfermedad en la mayoría de los pacientes dará lugar a complicaciones como resultado de citopenias y esplenomegalia; es decir, anemia, hemorragias, infecciones recurrentes, etc. En la práctica general, se debe iniciar el tratamiento si se presenta alguno de los criterios enumerados en la Tabla 2.Cuando se tome la decisión de no iniciar el tratamiento, se realizará una monitorización clínica y de laboratorio cada tres meses durante el primer año y cada seis meses a partir de entonces.,El tratamiento con HCL no se considera curativo, pero las estrategias de tratamiento actuales son capaces de alcanzar remisiones prolongadas, aumentando así las tasas de supervivencia global (algoritmo 1).36-39
los Criterios para iniciar el tratamiento.
1. Esplenomegalia sintomática
2. Citopenia que involucra al menos uno de los siguientes:
-hemoglobina g/dL
-plaquetas ×109/L
-neutrófilos ×109/l
3., Infecciones severas
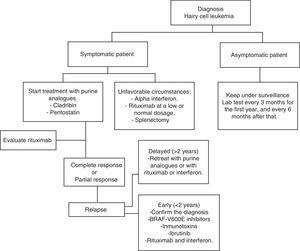
análogos de purina
en 1960, los hallazgos mostraron que el 30% de los niños con síndrome de inmunodeficiencia combinada grave carecían de la enzima deaminasa adenosina (ADA).,40 además, descubrieron que la acumulación de la forma de trifosfato de desoxiadenosina era responsable de la disminución de linfocitos.41 Teniendo en cuenta estas observaciones, se desarrollaron medicamentos capaces de unirse irreversiblemente a la ADA o antagonizar su acción. Después del tratamiento con análogos de purina, la acumulación de desoxiadenosina trifosfato da lugar a la ruptura y la inhibición de la reparación del ADN, lo que se traduce en apoptosis celular.,42
Pentostatina
también conocida como 2 ‘ – deoxicoformicina (dcF), un producto de Streptomyces antibioticus e inhibidores ADA, introducido por primera vez en 1980 como los primeros análogos de purina para el tratamiento de HCL. En la tabla 3,37,39,43,44 estudios específicos muestran tasas de respuesta a la pentostatina. El uso intravenoso de pentostatina en una proporción de 4mg/m2 una vez cada dos semanas hasta alcanzar la respuesta completa fue aprobado en los EE.UU. la Pentostatina es segura en pacientes con aligeramiento de creatinina > 60mL / min. Sin embargo, es necesaria una reducción de la dosis si esta depuración está entre 40 y 60 ml/min.,Se recomienda hidratación del paciente con 1,5 L de solución intravenosa con cada ciclo de pentostatina.30
La respuesta global varía entre el 88% y el 96%, mientras que la respuesta completa está entre el 44% y el 81%. Flinn et al. se estimó una tasa de supervivencia de 5 y 10 años de 90% y 81%, respectivamente, con una duración media de seguimiento de 9,3 años.La Pentostatina es generalmente bien tolerada y los efectos adversos más comunes son anemia, trombocitopenia y neutropenia.,46 sin embargo, se ha notificado que la pentostatina disminuye significativamente los recuentos de linfocitos CD4+ y CD8+, lo que podría aumentar las neoplasias malignas secundarias y la incidencia de infecciones.47,48
cladribina
se conoce como 2-clorodeoxiadenosina (CdA). En la tabla 4,49–52 Los estudios indican su eficacia. El esquema más utilizado consiste en Administrar 0,1 mg/kg/día en perfusión continua durante 7 días. En un estudio no aleatorizado, hubo pruebas que demostraban que no había diferencia estadísticamente significativa en los rangos de respuesta y toxicidad entre perfusiones (24h y 2h).,53 otro estudio aleatorizado comparó la administración diaria versus semanal de cladribina; no hubo hallazgos significativos en respuesta, supervivencia, tasas globales y de toxicidad.54 un estudio diferente mostró que el programa semanal redujo el riesgo de infecciones.55 una de las ventajas de la administración subcutánea es el hecho de que en la mayoría de los casos no requiere hospitalización. 0,14 mg / día durante 5 días tiene una tasa de respuesta del 95% 56, similar a la administración intravenosa. Los programas subcutáneos semanales tienen tasas de respuesta y toxicidad similares a las diarias.,57 con un solo ciclo de cladribina, se puede obtener una respuesta global de hasta el 100%, y las tasas de respuesta total difieren del 77% al 95%.49-52 Jehn y otros reportó una supervivencia global a los 12 años del 79%.36 en general, la cladribina es bien tolerada, siendo las citopenias y la fiebre los efectos adversos más comunes.
los Estudios sobre la eficacia de la cladribina en HCL casos.,
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days |
77.3 | 18.,6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days |
88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days |
95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.,1mg/kg/d 7 days |
79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence., Aun así, existen estudios retrospectivos que demuestran que ambos fármacos tienen una eficacia similar, en términos de respuesta completa y supervivencia libre de enfermedad.
otros tratamientos
los análogos de purina siguen siendo la primera línea de tratamiento, pero los nuevos descubrimientos relacionados con la fisiopatología del HCL han llevado a la creación de fármacos con diferentes dianas terapéuticas. Estos medicamentos están bajo investigación y algunos han mostrado resultados prometedores.
Rituximab
dado que el HCL es una neoplasia maligna de células B, es lógico usar un anticuerpo monoclonal contra CD20, como rituximab., Empleado como un fármaco independiente, rituximab puede alcanzar tasas de respuesta total de 10-54% en pacientes con una recaída de HCL, a una dosis de 375 mg/m2 una vez a la semana durante 4-8 semanas.58,59 Else et al. se revisaron retrospectivamente 18 pacientes que fueron tratados con análogos de purina en combinación con rituximab como segunda línea de tratamiento, después de haber sido tratados con análogos de purina en monoterapia. Todos los pacientes respondieron, con una tasa de respuesta completa del 89%.60
Rituximab a 375 mg / m2 por semana durante 8 semanas como tratamiento inicial después de la administración 5.,6 mg/m2 de cladribina mediante perfusión IV de 2 horas durante 5 días genera una respuesta total del 100%.61 en situaciones especiales o desfavorables, 100mg de rituximab por semana puede ser utilizado durante 4-6 semanas. Esto es menos costoso y generalmente es efectivo, especialmente cuando se combina con interferón.
mientras que los análogos de purina pueden no ser capaces de eliminar el HCL, ya que la enfermedad residual mínima (ERM) detectada después de la administración de cladribina siempre es fuertemente CD20+, la erradicación de la ERM se puede obtener utilizando rituximab. Rivandi et al., se demostró en un estudio preliminar que el rituximab a dosis convencionales durante un período de 8 semanas funciona con gran actividad, eliminando la ERM en 13 pacientes, cuando se usa 4 semanas después de la administración de cladribina.62
Vemurafenib
Como se describió anteriormente, la mutación BRAF V600E es la clave genética en el HCL. Por lo tanto, es una diana terapéutica que se ha estudiado en los últimos años. Vemurafenib es un inhibidor oral de BRAF V600E. Tiacci et al., se realizó un estudio para medir la actividad y seguridad de Vemurafenib en pacientes con CPH que recayeron después del tratamiento con análogos de purina o que fueron refractarios a la administración de análogos de purina. La tasa de respuesta Global fue de 96% y la tasa de respuesta completa fue de 35%, con una supervivencia sin recaída media de 19 meses. Los efectos adversos fueron erupción, artralgia y artritis.
dado que se ha observado una relación positiva entre el uso de Vemurafenib y la aparición de neoplasias dermatológicas, se recomiendan exploraciones frecuentes de la piel.,
Ibrutinib
un inhibidor selectivo e irreversible de la tirosina quinasa de Burton interviene en la vía de señalización de las células B.64 recientemente se ha iniciado un ensayo clínico de ibrutinib en pacientes con recaída del HCL. Los datos preliminares de eficacia y seguridad muestran efectos adversos como erupciones, diarrea y artralgia. Este ensayo clínico se está llevando a cabo actualmente en varios centros en los EE.UU. (NCT01841723).
Inmunotoxinas
con el fin de aumentar la citotoxicidad de anticuerpos monoclonales, se han creado técnicas que facilitan la producción de conjugados anticuerpo–toxina o anticuerpo–fármaco., Una inmunotoxina es la fusión entre una toxina bacteriana (es decir, Pseudomonas exotoxina o difteria) y la fracción variable de un anticuerpo monoclonal, cuyo objetivo específico se encuentra en la superficie de células neoplásicas como CD25 o CD22. Esta toxina se libera en el interior de la célula neoplásica e interfiere con la síntesis de proteínas.65
BL22 es una inmunotoxina contra CD22 fusionada con una forma truncada de la P. exotoxina PE38. En un ensayo clínico de fase II, se probó BL22 en 36 casos de recaída o enfermedad refractaria al HCL., Después de un ciclo (40 mg/kg cada dos días, tres dosis), la tasa de respuesta completa fue del 25% y la tasa de respuesta global fue del 50%. Estas respuestas mejoraron a una tasa de respuesta total del 47% y una tasa de respuesta global del 72% después de la repetición del tratamiento (solo en pacientes con citopenias). Dos pacientes desarrollaron síndrome hemolítico urémico sin necesidad de recurrir a plasmaféresis.Posteriormente, se desarrolló moxetumomab pasudotox como una versión modificada de BL22 con mayor afinidad y citotoxicidad., En un ensayo de fase I, que incluyó a 28 pacientes con recidiva y resistencia al HCL, se obtuvo una tasa de respuesta global del 86%, incluida una tasa de respuesta completa sostenida en el 46% de los pacientes.67
opciones terapéuticas en circunstancias desfavorables
aunque el HCL se trata en la mayoría de los países desarrollados con cladribina y pentostatina, es un hecho que estos medicamentos no solo son caros, sino que no están disponibles en México y en muchos países con recursos limitados., Por lo tanto, en este tipo de circunstancias, hay otras opciones terapéuticas asequibles con resultados favorables
el interferón alfa para el tratamiento de pacientes con HCL se introdujo por primera vez en 1984. Hoy en día, su uso es limitado, principalmente debido a la gran eficacia de los análogos de purina. Por otro lado, en países con bajos recursos económicos, es una opción barata y que ha demostrado resultados similares a los de la cladribina en términos de supervivencia global. Ruiz-Delgado et al., se realizó un estudio comparativo entre interferón alfa (n = 18) y cladribina (n=11), en el que la diferencia en la supervivencia global entre ambos grupos no fue estadísticamente significativa; el 94% a los 217 meses en el grupo de interferón y el 91% a los 133 meses en el grupo de cladribina.68 en un estudio realizado en nuestro centro, nueve pacientes con HCL recibieron tres mega – unidades de IFN tres veces por semana durante 12 semanas, posteriormente recibieron tratamiento una vez más durante 8 semanas cuando hubo una reactivación de la leucemia o después de 10 meses de observación cada año., Todos los pacientes tuvieron remisión hematológica antes de las 12 semanas de tratamiento. Esta opción terapéutica es más barata, efectiva y comparable a otras formas de terapia con IFN en el tratamiento y mantenimiento de pacientes con este tipo de leucemia.69 es posible combinar interferón con rituximab sin aumentar los efectos tóxicos y mejorar la eficacia.
la esplenectomía fue la primera intervención que cambió significativamente la supervivencia de los pacientes. Hoy en día, rara vez se usa., Puede recomendarse en pacientes con esplenomegalia masiva dolorosa (> 10 cm bajo el borde costal) y con infiltración mínima de la médula ósea, o en pacientes refractarios al tratamiento con análogos de interferón y purina.33 estudios retrospectivos muestran una tasa de respuesta completa de 40-62%, y una tasa media de supervivencia a 5 años de hasta 68%.70,71 Lad et al. publicado un estudio retrospectivo, que incluyó 24 pacientes con diagnóstico de HCL, se dividieron en dos grupos: 17 pacientes recibieron cladribina y 7 fueron esplenectomizados., 75% de los pacientes en el grupo de esplenectomía mostraron remisión total, 94% lo hicieron en el grupo de cladribina. Un hallazgo interesante al comparar ambos grupos fue que no se observaron diferencias estadísticamente significativas en cuanto a la supervivencia sin leucemia y la supervivencia global.
pronóstico
El Tiempo de supervivencia en los pacientes después del diagnóstico fue de 4 años antes de que se conociera el tratamiento, debido a complicaciones derivadas de citopenias, incluyendo hemorragias e infecciones. A partir de entonces, con la esplenectomía como tratamiento de primera línea, hubo una respuesta completa de 40-62% y una tasa de supervivencia de 5 años de 61-68%., Luego, el interferón alfa se utilizó como el primer fármaco con beneficios en el tratamiento del HCL. Sin embargo, su tasa de respuesta completa fue baja, del 10%.
hoy en día, con análogos de purina (pentostatina y cladribina), se induce una respuesta completa hasta el 80% de los pacientes con una mediana de supervivencia de 10 años. Las tasas de respuesta Global son del 96-100%, con una tasa de respuesta completa del 80% y una tasa de supervivencia de 10 años que oscila entre el 85% y el 100%.74 a pesar de ello, una proporción significativa de pacientes con HCL fallan en su respuesta al tratamiento o se vuelven resistentes., Hasta el 48% de los pacientes recaen en los siguientes 15 años.75 el futuro de los pacientes con HCL es muy favorable. El desafío es identificar esta malignidad lo antes posible y tratarla adecuadamente utilizando los recursos disponibles.
conflicto de intereses
los autores no tienen conflictos de intereses que declarar.