en el post anterior presentamos a los directores de Orto ,para y meta en sustitución aromática electrofílica. Anteriormente cubrimos el mecanismo de sustitución aromática electrofílica, y demostramos que el mecanismo procede a través de un intermediario carbocatiónico.,
Hoy, vamos a unir esos dos conceptos, y tratar de mostrar que si un sustituyente es un orto-, para – o meta – director depende de cómo el sustituyente afecta la estabilidad del intermedio carbocatión.
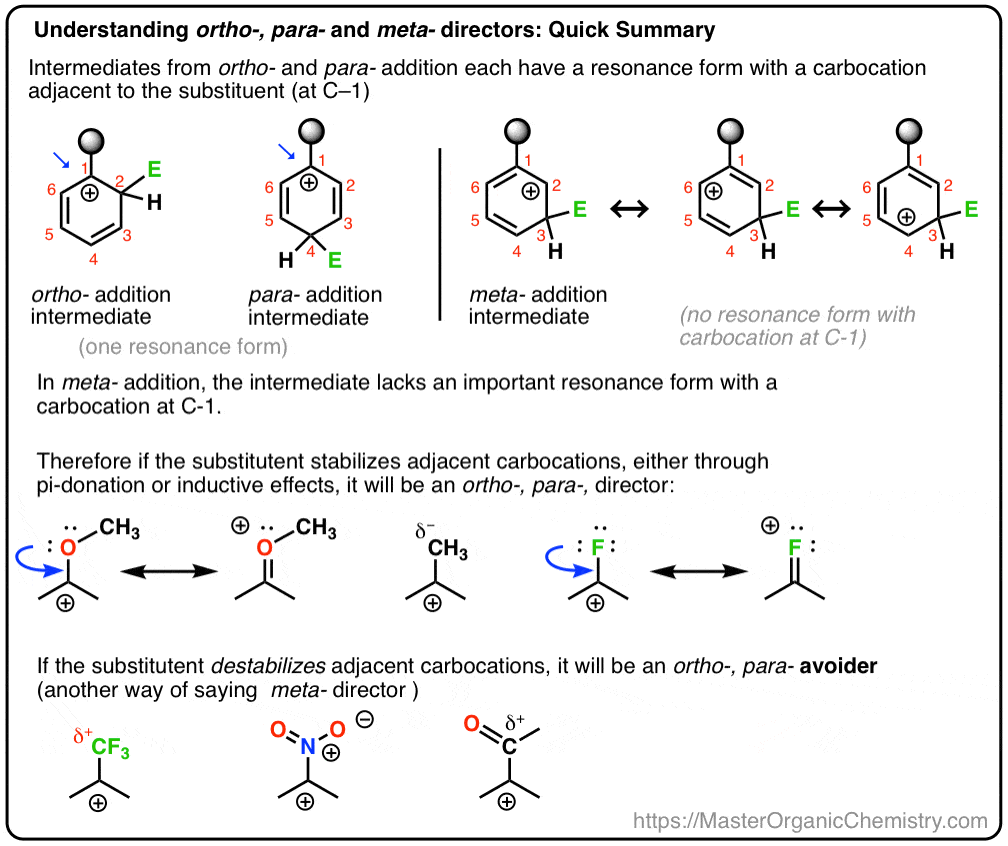
y también veremos por qué un nombre alternativo para meta-director podría ser, «ortho -, para-avoider». :- )
tabla de contenidos
- Las Carbocaciones se estabilizan por átomos adyacentes con pares solitarios
- Estudio de Caso #1: un ortho -, para-Director (OCH3)
- Sí, el oxígeno es electronegativo., ¡Pero La Donación De Su Par Solitario Lo Compensa Con Creces!
- El diagrama de reacción-energía para un director de Ortho para
- Estudio de Caso # 2: un meta-director (CF3).
- lo que llamamos » meta-directores «son más como»ortho-, para– Avoiders»
- El diagrama de energía de reacción para un meta – Director
- Entonces, ¿Por qué los halógenos son Ortho -, Para – directores?
- Notas
- Prueba a Ti mismo!,
- referencias (avanzadas) y lecturas adicionales
las Carbocaciones son estabilizadas por átomos adyacentes con pares solitarios
pensemos en qué factores aumentan la estabilidad de las carbocaciones.
Las Carbocaciones son especies pobres en electrones con seis electrones en su capa de Valencia. Por lo tanto, cualquier sustituyente que puede donar densidad de electrones hacia el carbocation se estabilizará. Esto incluye:
- sustituyentes liberadores de electrones que pueden actuar como» sigma-donadores » (E. G., muchos grupos alquilos, que donan densidad electrónica a través de efectos inductivos )
- átomos adyacentes con pares solitarios que pueden actuar como «donantes pi»a la carbocación
- enlaces pi c–C adyacentes que pueden estabilizar la carbocación a través de la deslocalización de carga («estabilización de resonancia», en otras palabras).
y factores que desestabilizan las carbocaciones?
- cualquier sustituyente que elimina la densidad de electrones de una carbocación, como los grupos fuertes de retiro de electrones.
así que aquí hay un quiz rápido., Teniendo en cuenta todo lo escrito anteriormente, ¿cuál de las dos carbocaciones a continuación será más estable?
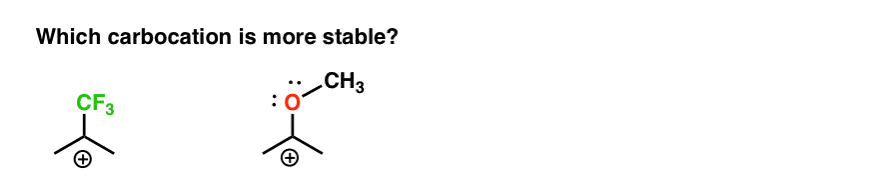
si puede responder bien a esta pregunta, estará en la mayor parte del camino hacia la comprensión de la diferencia entre orto-, para – y meta – directores.
examinemos dos casos de estudio. Veremos una reacción de sustitución aromática electrofílica genérica del benceno con un orto -, para-director (metoxibenceno) y examinaremos los intermedios que se obtienen del ataque en las posiciones Orto, meta y para., Luego realizaremos el mismo ejercicio con un meta-director (trifluorometilbenceno).
en el proceso, esperamos entender por qué el metoxibenceno favorece a los productos ortho y para, mientras que el trifluorometilbenceno favorece al meta – producto.
Estudio de Caso #1: un ortho-,para – Director (OCH3)
sustitución aromática electrofílica de metoxibenceno tiende a dar ortho – y para – productos con muy poco meta- ., Esto es lo que sucede en la nitración, por ejemplo:
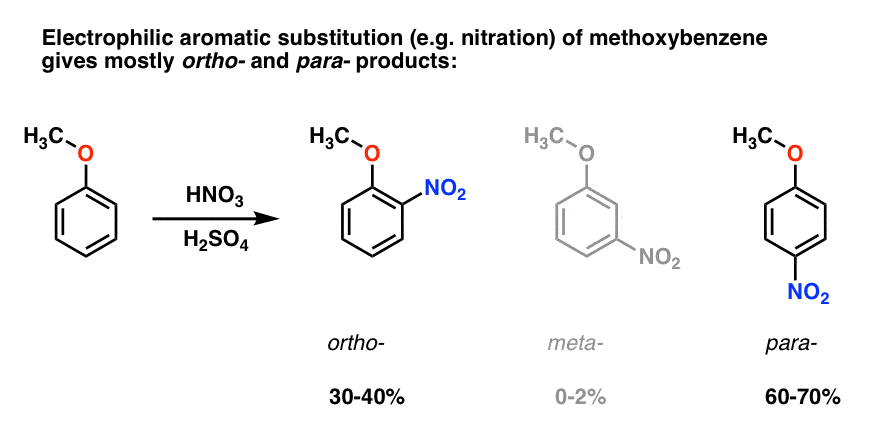
El para – producto domina (60-70%), con ortho – cerca detrás (30-40%) y solo un rastro de la meta– .
¿por Qué?
3. Sí, El Oxígeno Es Electronegativo. ¡Pero La Donación De Su Par Solitario Lo Compensa Con Creces!
Aquí está el camino que conduce al Orto-producto., Rompemos un enlace pi C–C y formamos un nuevo enlace C–E (donde E está destinado a representar un electrofilo genérico), generando una carbocación estabilizada por resonancia:
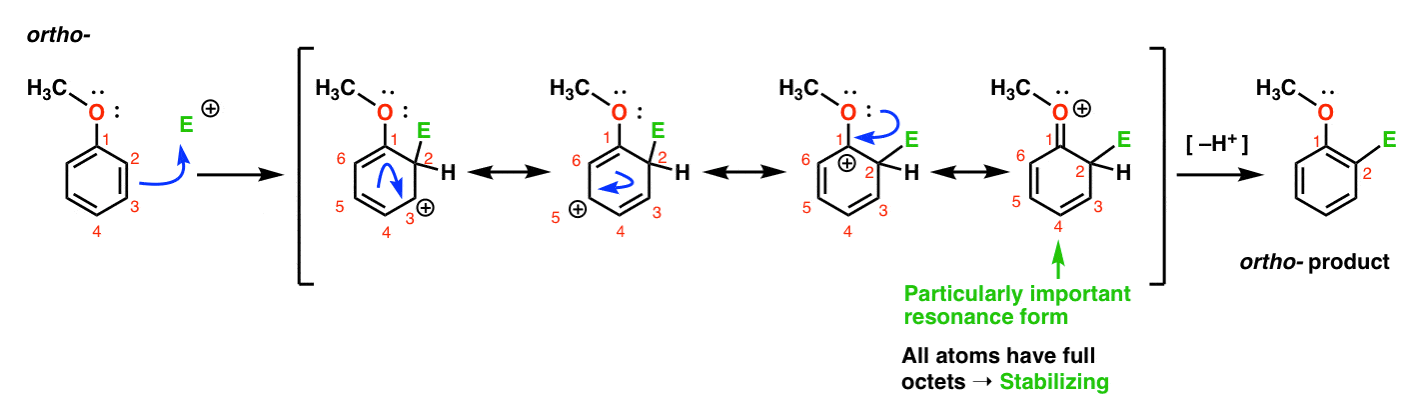
Si miras de cerca, debes tener en cuenta que podemos formar cuatro formas de resonancia . Tres de ellos tienen carbocaciones completas (en C1, C3 y C5), pero es el cuarto – la forma de resonancia donde hay un enlace C=o pi – lo que es particularmente importante. ¿Por qué? En esta forma de resonancia, todos los átomos de carbono tienen un octeto completo de electrones., Eso es porque el oxígeno directamente unido al anillo puede donar un par solitario a la carbocación adyacente, formando un enlace pi. Este es un ejemplo de donación de pi.
Aquí está la vía para el ataque del electrófilo en la meta – posición. Nota la diferencia!
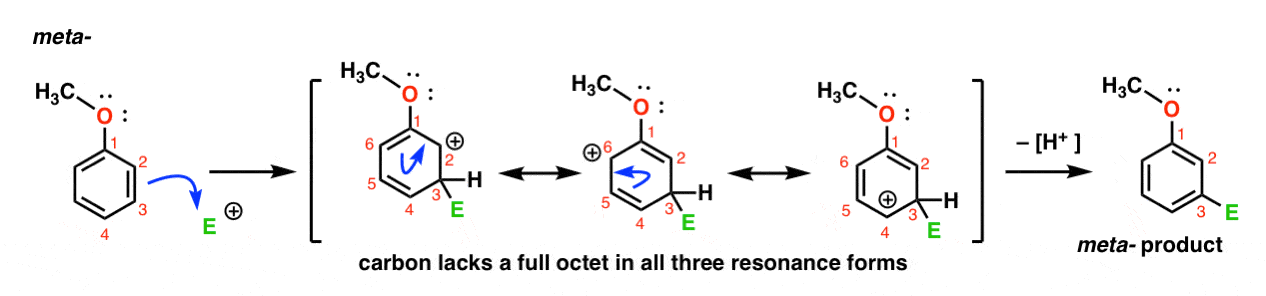
El ataque del electrófilo en C-3 resulta en una carbocación que puede deslocalizarse a través de resonancia a C2, C4 y C6. Sin embargo, no hay manera de «mover» la carbocación a C1, lo que significa que no hay una forma de resonancia razonable donde todos los átomos tengan octetos completos.,
esto hace que el intermedio meta – carbocation sea mucho menos estable que el intermedio Orto-carbocation.
finalmente, aquí está la reacción que conduce al para – producto:
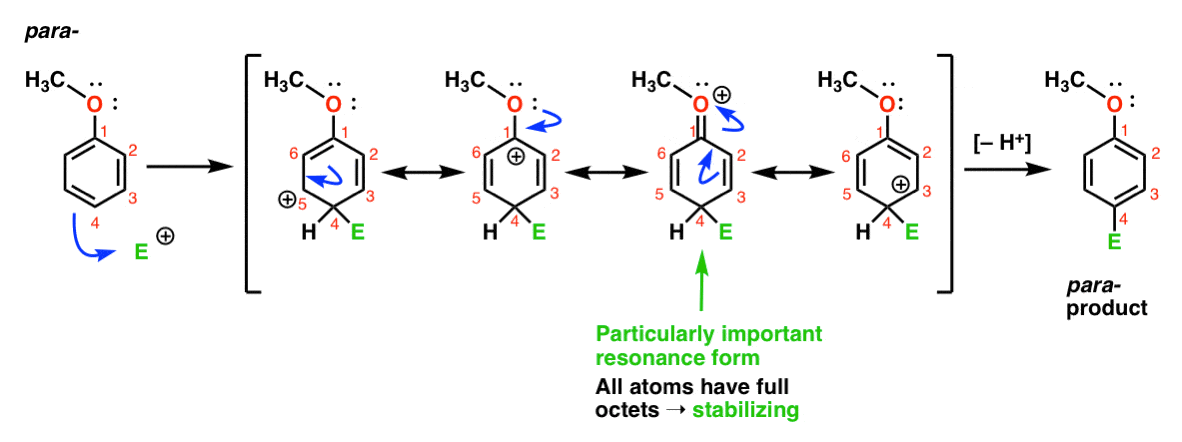
Aquí podemos dibujar nuevamente formas de resonancia con carbocaciones en C1, C3 y C5, así como una cuarta forma de resonancia donde el átomo de oxígeno Unido dona un par de electrones a la carbocación en C1, lo que resulta en un octeto completo en carbono. Esta es una situación esencialmente el mismo como el orto – intermedio.,
por lo tanto, mediante el análisis de las formas de resonancia (y marque mi palabra, es posible que se le pida que haga lo mismo en una prueba en un futuro cercano) podemos ver que las carbocaciones intermedias resultantes de la orto – y para– adición son considerablemente más estables que las intermedias de la meta– adición.
esto explica por qué los productos ortho y para dominan, y el meta-producto es menor.
Podría ser útil visualizar esto dibujando un diagrama de energía de reacción.
4., El diagrama de energía de reacción para un director de Orto para
el estado de transición que conduce a los productos de Orto – y para-adición es mucho más bajo en energía que el meta– .
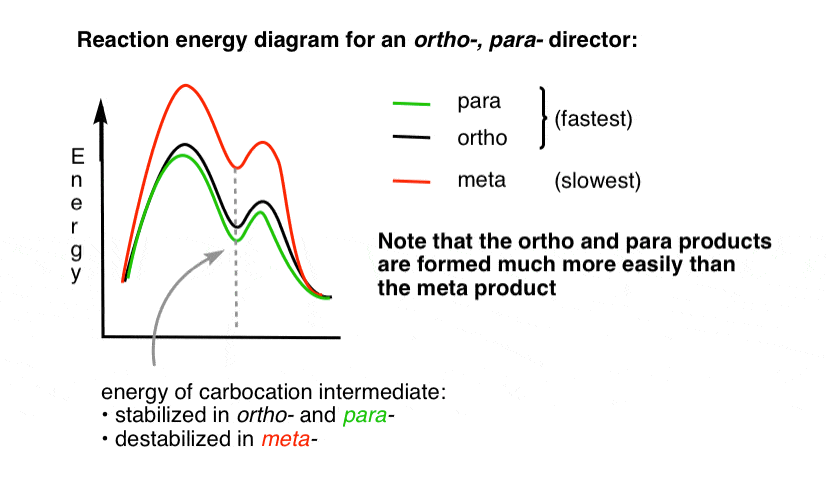
una pregunta – ¿por qué crees que para – podría ser favorecido (por ejemplo, 60% -70% de rendimiento para el para– producto de nitración, arriba, frente a 30-40% para orto-) a pesar de que hay dos Orto – posiciones y solo un para- ?
piense en eso por un minuto, y lo abordaremos a continuación.
5. Estudio de Caso # 2: un meta-director (CF3).
entonces, ¿qué pasa con CF3?, ¿Por qué da meta-productos? Hagamos el mismo tipo de análisis.
Aquí está el camino que conduce al Orto-producto. Al igual que con el ortointermedio anterior, podemos dibujar tres formas de resonancia colocando el carbocatión en C1, C3 y C5, respectivamente.
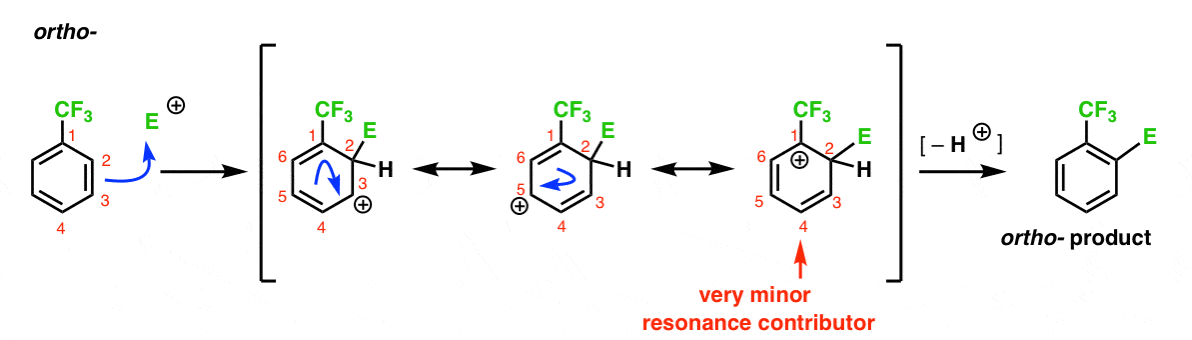
Así que, ¿qué es diferente en este caso?
lo que es diferente es la presencia de un grupo fuerte de retiro de electrones (CF3) en C1, y esto cambia completamente el juego de pelota.
como hemos dicho, las carbocaciones son desestabilizadas por vecinos que retiran la densidad de electrones., Por lo tanto, esperaríamos que esto sea un contribuyente de resonancia muy menor, con el resultado de que la carga positiva solo se deslocaliza sobre C3 y C5.
hemos observado que CF3 es un meta-director. Es razonable pensar que hay algo en el meta – que lo hace particularmente estable. Al analizar el intermedio para el meta– producto a continuación, ¿puede ver por qué?
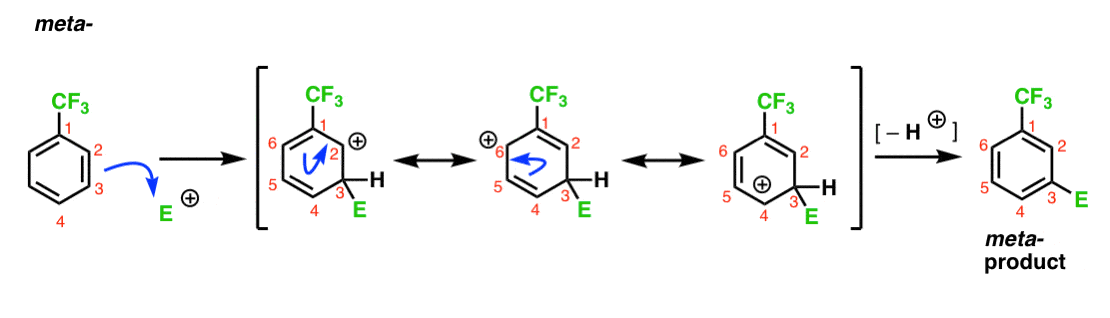
Hmmm. No hay intermedio donde un carbono tenga un octeto completo. No hay formas de resonancia particularmente estables.
es una especie de «meh», ¿no?
6., Lo que llamamos «meta– directores» son más como «ortho-, para– Avoiders»
por otro lado, tampoco hay formas de resonancia particularmente inestables. Dado que la carga positiva se localiza en C2, C4 y C6, se evita una situación en la que la carga positiva sea directamente adyacente al grupo CF3 que retira electrones. Y la carga positiva se deslocaliza muy bien a lo largo del anillo, a diferencia de la Orto – situación anterior.
así que es un intermedio «menos malo» que el ortho-.,
finalmente, el para – intermedio tiene el mismo problema que el orto -; un intermedio con una carga positiva en C1, adyacente al CF3.
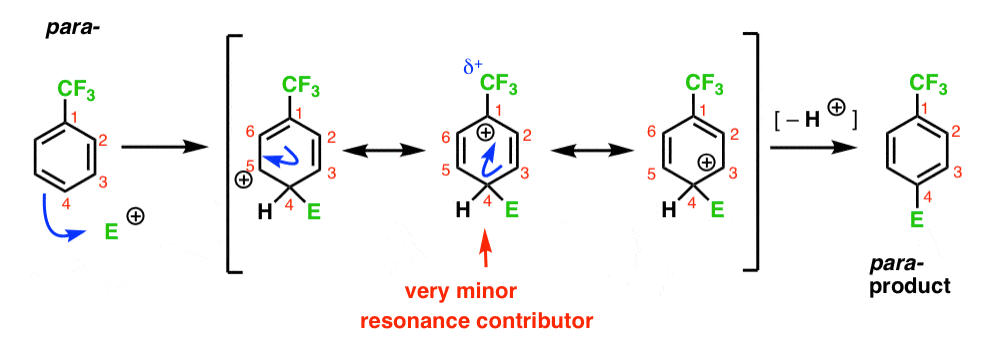
esto da como resultado solo dos formas de resonancia importantes, lo que conduce a un intermedio de carbocación menos deslocalizado (y por lo tanto menos estable).
En pocas palabras: el meta carbocation intermedio es más estable que los orto – o los PARA-intermedios.
pero no es porque el sustituyente en sí tiene algún efecto estabilizador en el meta– intermedio. .,
yo diría que es más útil pensar en el meta – como menos inestable que el orto – y para– .
de esta manera, no es tanto que CF3 sea un meta – director, tanto que sea un «ortho– , para– avoider». «
7. El Diagrama de Energía de Reacción Para Un meta – Director
he Aquí un bosquejo del diagrama de energía de reacción:
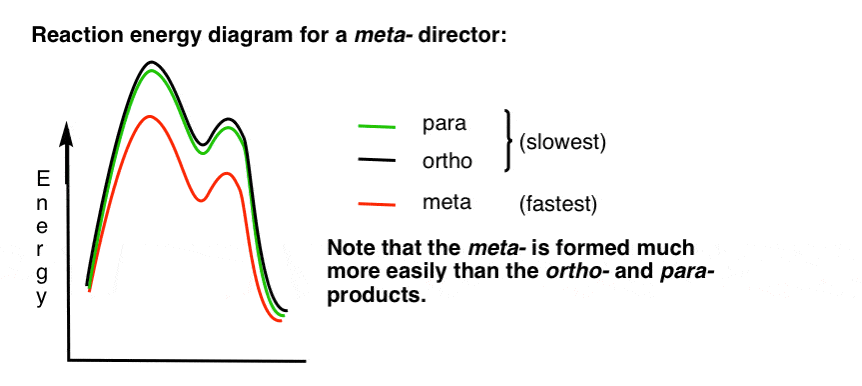
Bien.
de vuelta a nuestra pregunta original. Así que carbocation es más estable?,
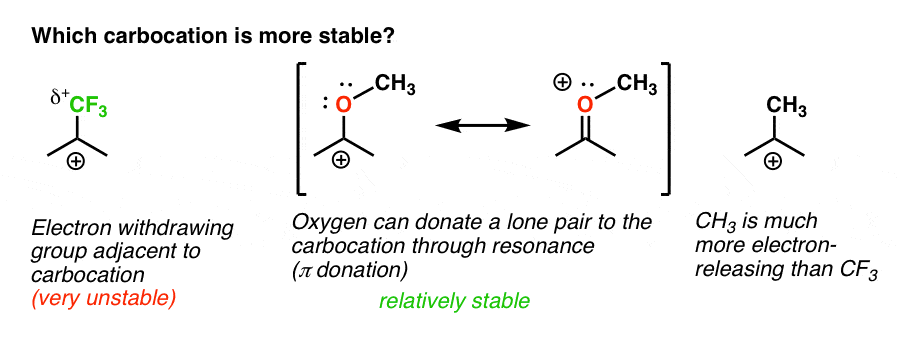
claramente, la carbocación con un oxígeno adyacente que lleva un par solitario es mucho más estable que una carbocación adyacente a un grupo de extracción de electrones como CF3.
y eso comprende la diferencia entre un orto-, para – director como OCH3 y un meta – director como CF3.
vale la pena señalar que la mayoría de los grupos alquilos (como CH3 ) carecen de un par solitario, pero siguen siendo orto-, para – directores., Esto es consistente con todo lo que hemos aprendido antes sobre cómo los grupos alquilos generalmente se estabilizan para carbocaciones-recordemos que la estabilidad de carbocación generalmente aumenta con la sustitución
así que sobre esos para-productos
¿Por qué se producen para – productos a una tasa mayor que Orto- ?
la respuesta es un obstáculo estérico.
El Ataque en la posición para está menos ocupado por el sustituyente que el ataque en las posiciones Orto.
entonces, ¿por qué los halógenos son Ortho -, Para-directores?
esto nos deja con el ejemplo algo complicado de los halógenos.,
¿Por qué el flúor, el cloro, el bromo y el yodo son orto -, para – directores a pesar de que están desactivando grupos?
la respuesta no debería ser una sorpresa si has estado siguiendo, pero vamos a dejar esto para el próximo post, porque este ya es suficiente.
siguiente post: ¿por qué son los halógenos ortho -, para-directores?
notas
Nota 1. Es más correcto decir que los orto – y para – productos dominan porque los estados de transición que conducen a estos productos son más bajos en energía, en lugar de las energías de los intermedios mismos., Después de todo, es la energía de los estados de transición la que determina la barrera de activación, y por lo tanto la velocidad de reacción.
Nota 2. O hiperconjugación, que la mayoría de los libros de texto (con la notable excepción de Maitland Jones) generalmente evitan.
¡Hazte una prueba tú mismo!,/div>
 haga Clic en Voltear
haga Clic en Voltear
haga Clic en Voltear  haga Clic en Voltear
haga Clic en Voltear 
(Avanzado) Referencias y lecturas Adicionales
- A., F. Holleman, Die direkte Einführung von Substituenten in den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456 DOI: 10.1002/recl.19100291205
A. F Holleman de 1910 dijo que la orientación ortho–para está asociada con la activación y la orientación meta con la desactivación. - – La naturaleza del efecto alterno en las cadenas de carbono. Part XXIII. Anomalous orientation by halogens, and its bearing on the problem of the ortho–para ratio, iN aromatic substitution
Christopher Kelk Ingold and Charles Cyril Norrey Vass
J. Chem. Soc. 1928, 417-425 DOI: 10.,1039 / JR9280000417
este artículo discute los efectos de dirección en 1,2-dihalobencenos. - Algunas observaciones en relación con el impedimento estérico y los efectos de los sustituyentes en el orto : párr relación en substitución aromática
P. B. D. de la Mare
J. Chem. Soc. 1949, 2871-2874
DOI: 10.1039/JR9490002871
en este artículo, de la Mare discute varios factores que pueden explicar un predominio de Para sobre la selectividad de Orto, después de dividir el rendimiento del producto de orto por 2 (ya que hay 2 posiciones de Orto pero solo 1 Posición de para en los bencenos monosustituidos)., - Volume effects of alkyl groups in aromatic compounds. Parte V. La monosulfonación de P-cimeno R. J. W. Le Fèvre J. Chem. Soc. 1934, 1501-1502
DOI: 10.1039 / JR9340001501
en P-cimeno, el producto principal obtenido sobre la sulfonación electrofílica es el producto 2 (orto al grupo metilo), probablemente debido a las estéricas. - Effects of Alkyl Groups in Electrophilic Additions and Substitutions
COHN, H., HUGHES, E., JONES, M. and PEELING, M. G.
Nature 1952, 169, 291
DOI: 1038/169291a0
Este artículo tiene datos comparando la nitración de T-butilbenceno y tolueno., El T-butilbenceno es mucho más P-Director que el tolueno (79.5% para para el T-butilbenceno vs. 40% para para el tolueno), que es probablemente debido a sterics (Orto enfoque es bloqueado por el Grupo T-butil más voluminoso). - Distribution of Isomers in the Mononitration of Ethyl – and Isopropylbenzene. Further Evidence for a Steric Effect in Isomer Distribution Herbert C. Brown and W. Hallam Bonner Journal of the American Chemical Society 1954, 76 (2), 605-606 DOI: 10.,1021 / ja01631a084
El Cuadro II de este documento ilustra que el producto ortho obtenido de la nitración de monoalquilbencenos disminuye a medida que el grupo alquilo se hace más grande (por ejemplo, el T-butilbenceno produce muy poco producto ortho tras la nitración en comparación con el tolueno). - – La naturaleza del efecto alterno en las cadenas de carbono. Part XXII. An attempt further to define the probable mechanism of orientation in aromatic substitution
Christopher Kelk Ingold and Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926 DOI: 10.,1039 / JR9270002918
Un artículo temprano del influyente químico físico orgánico, Prof. C. K. Ingold, afirmando que los halogenobencenos son inductivamente retiradores de electrones pero simultáneamente estabilizadores de resonancia. - the Anomalous Reactivity of Fluorobenzene in Electrophilic Aromatic Substitution and Related Phenomena
Joel Rosenthal and David I., Schuster
Journal of Chemical Education 2003, 80 (6), 679
DOI: 1021/ed080p679
Un artículo muy interesante, adecuado para estudiantes curiosos, y discute algo que la mayoría de los químicos orgánicos practicantes sabrán empíricamente: el fluorobenceno es casi tan reactivo como el benceno en las reacciones EAS o Friedel-Crafts, ¡lo cual es contraintuitivo cuando uno considera los efectos electrónicos! - Organic chemistry
N. Haworth, C. K. Ingold, T. A. Henry
Ann. República Prog. Chem. 1926, 23, 74-185 DOI: 10.1039/AR9262300074 An early paper discussing directing effects in EAS. Pp., 136 y 137 contienen figuras que representan el flujo de electrones. Pg. 140 discute meta-dirección, que ocurre por las razones’ opuestas ‘ como O / P-dirección por grupos alquilos.