dans le post précédent, nous avons présenté les directeurs ortho, para et méta – dans la substitution électrophile aromatique. Auparavant, nous avons couvert le mécanisme de substitution aromatique électrophile et montré que le mécanisme passe par un intermédiaire carbocation.,
Aujourd’hui, nous allons lier ces deux concepts ensemble, et essayer de montrer que si un substituant est un ortho-, para – ou méta – directeur dépend de la façon dont le substituant affecte la stabilité de l’intermédiaire carbocation.
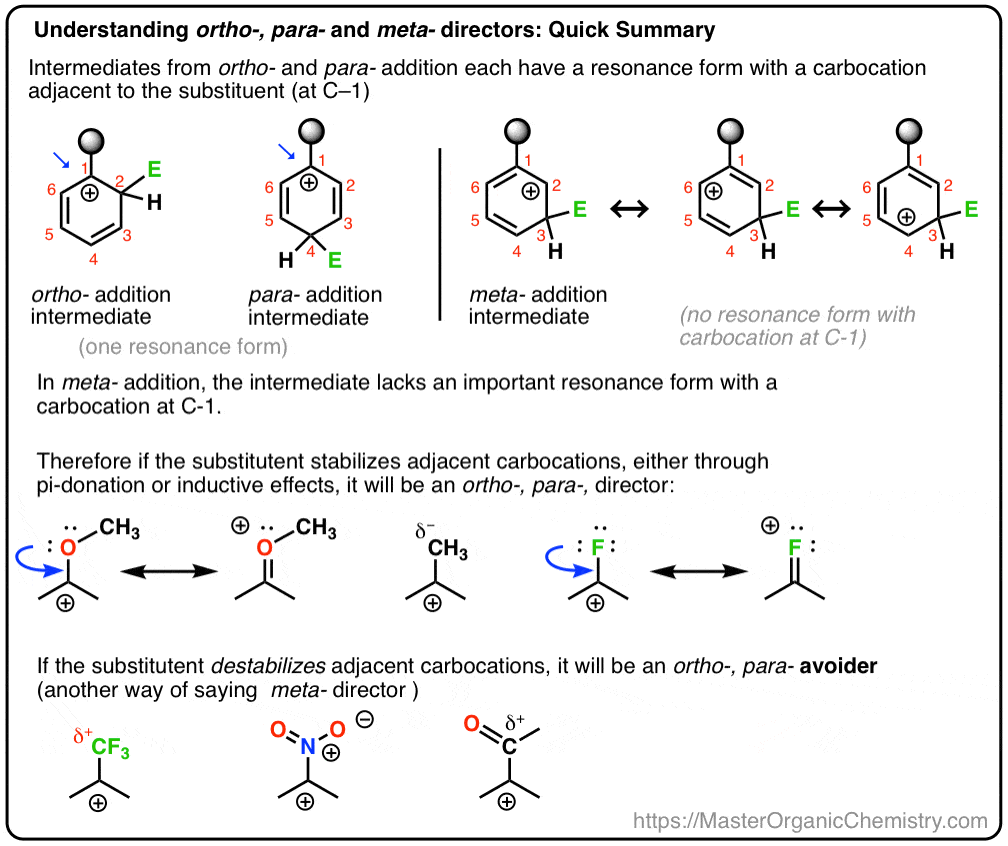
Et nous allons aussi voir pourquoi un nom alternatif pour la méta – directeur pourrait être, « ortho-, para – avoider”. :- )
table des matières
- Les Carbocations sont stabilisés par des atomes adjacents avec des paires isolées
- étude de Cas #1: un ortho -, para-Directeur (OCH3)
- Oui, L’oxygène est électronégatif., Mais Le Don De Sa Paire Solitaire Compense Plus Que Cela!
- Le diagramme de réaction-énergie pour un directeur Ortho Para
- étude de Cas #2: un méta – directeur (CF3).
- ce que nous appelons » méta-directeurs « ressemble plus à”ortho-, para– Avoiders »
- Le Diagramme D’énergie de réaction pour un méta – Directeur
- alors pourquoi les halogènes sont-ils Ortho -, Para-directeurs?
- Notes
- testez-Vous!,
- (avancé) références et lectures complémentaires
les Carbocations sont stabilisés par des atomes adjacents avec des paires isolées
réfléchissons aux facteurs qui augmentent la stabilité des carbocations.
Les Carbocations sont des espèces pauvres en électrons avec six électrons dans leur coquille de valence. Ainsi, tout substituant qui peut donner une densité électronique vers le carbocation se stabilisera. Cela inclut:
- substituants libérant des électrons qui peuvent agir comme « donneurs de sigma » (par exemple, de nombreux groupes alkyles, qui donnent la densité électronique par des effets inductifs )
- atomes adjacents avec des paires isolées qui peuvent agir comme « donneurs de pi” au carbocation
- liaisons C–C Pi adjacentes qui peuvent stabiliser le carbocation par délocalisation de charge (« stabilisation par résonance”, en d’autres termes).
Et les facteurs qui déstabilisent les carbocations?
- tout substituant qui enlève la densité électronique d’un carbocation, tel que les groupes électron-retirants forts.
Voici donc un quiz rapide., Compte tenu de tout ce qui est écrit ci-dessus, lequel des deux carbocations ci-dessous sera le plus stable?
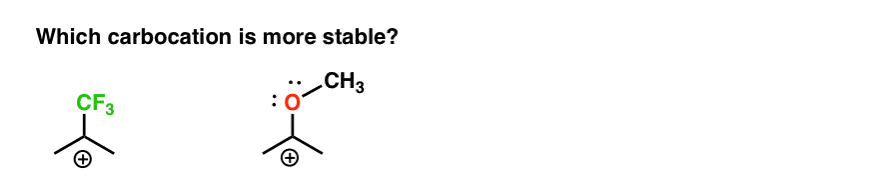
Si vous pouvez bien répondre à cette question, vous êtes la plupart du chemin vers la compréhension de la différence entre ortho-, para – et méta – directeurs.
examinons deux études de cas. Nous examinerons une réaction générique de substitution électrophile aromatique du benzène avec un ortho -, para-directeur (méthoxybenzène) et examinerons les intermédiaires obtenus par attaque aux positions ortho, méta et para., Ensuite, nous effectuerons le même exercice avec un méta-directeur (trifluorométhylbenzène).
dans le processus, nous espérons comprendre pourquoi le méthoxybenzène favorise les ortho – et para– produits tandis que le trifluorométhylbenzène favorise le méta – produit.
étude de Cas #1: un ortho-,para – Directeur (OCH3)
la substitution électrophile aromatique du méthoxybenzène tend à donner des ortho – et para – produits avec très peu de méta- ., Voici ce qui se passe en nitration, par exemple:
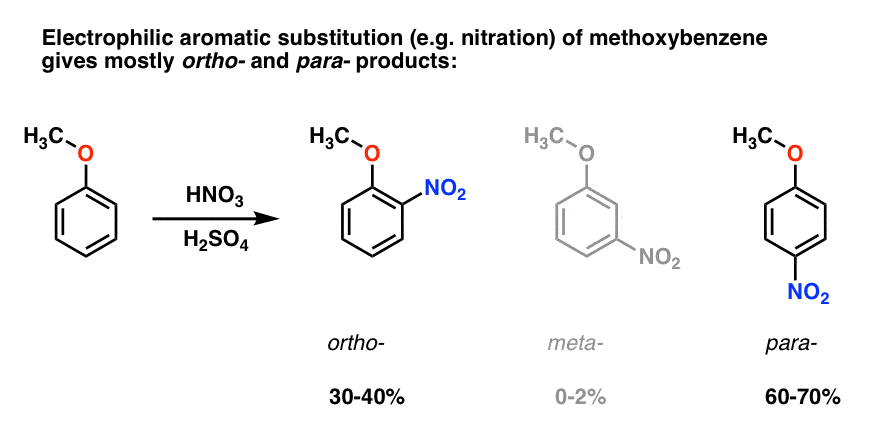
Le para – produit domine (60-70%), avec ortho – close derrière (30-40%) et seulement une trace du Méta– .
pourquoi?
3. Oui, L’Oxygène Est Électronégatif. Mais Le Don De Sa Paire Solitaire Compense Plus Que Cela!
Voici la voie menant à l’ortho – produit., Nous rompons une liaison C-C pi et formons une nouvelle liaison C-E (où E est destiné à représenter un électrophile Générique), générant un carbocation stabilisé par résonance:
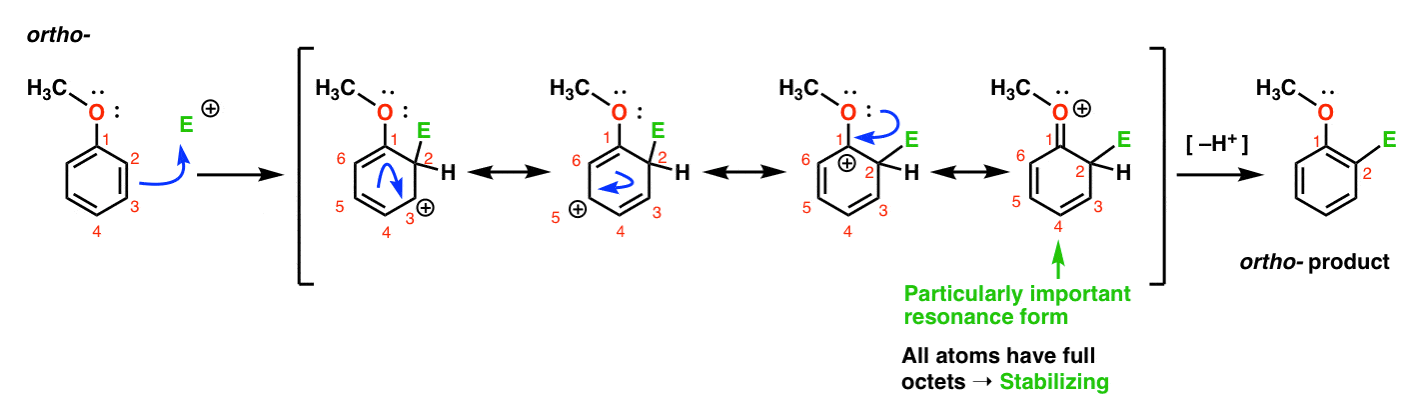
Si vous regardez attentivement, vous devriez noter que nous pouvons former quatre formes de résonance . Trois d’entre eux portent des carbocations complètes (sur C1, C3 et C5), mais c’est la quatrième – la forme de résonance où il y a une liaison C=O pi – qui est particulièrement importante. Pourquoi? Dans cette forme de résonance, tous les atomes de carbone ont un octet complet d’électrons., C’est parce que l’oxygène directement lié à l’anneau peut donner une paire solitaire au carbocation adjacent, formant une liaison pi. Ceci est un exemple de don de pi.
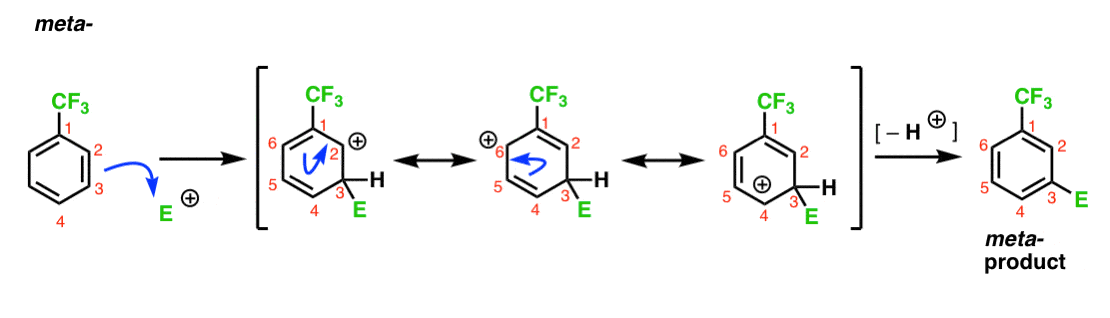
Voici la voie d’attaque de l’électrophile à la méta – position. Notez la différence!
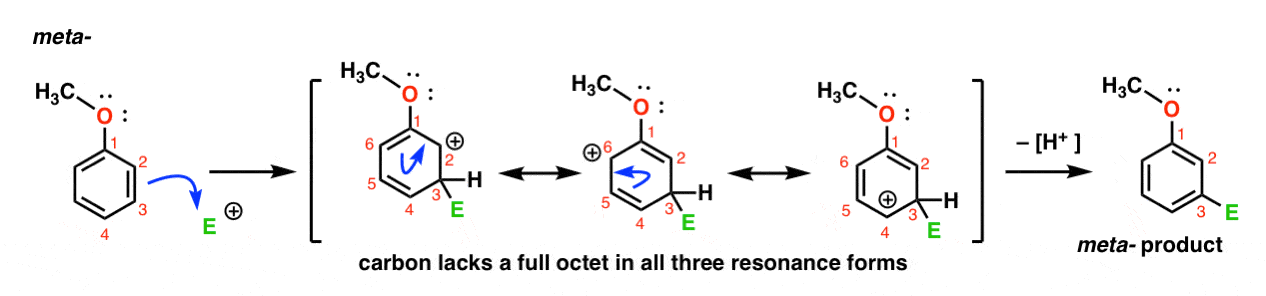
L’attaque de L’électrophile en C-3 entraîne un carbocation qui peut être délocalisé par résonance en C2, C4 et C6. Cependant, il n’y a aucun moyen de « déplacer” le carbocation vers C1, ce qui signifie qu’il n’y a pas de forme de résonance raisonnable où tous les atomes ont des octets complets.,
cela rend l’intermédiaire méta – carbocation beaucoup moins stable que l’intermédiaire ortho – carbocation.
enfin, voici la réaction menant au para – produit:
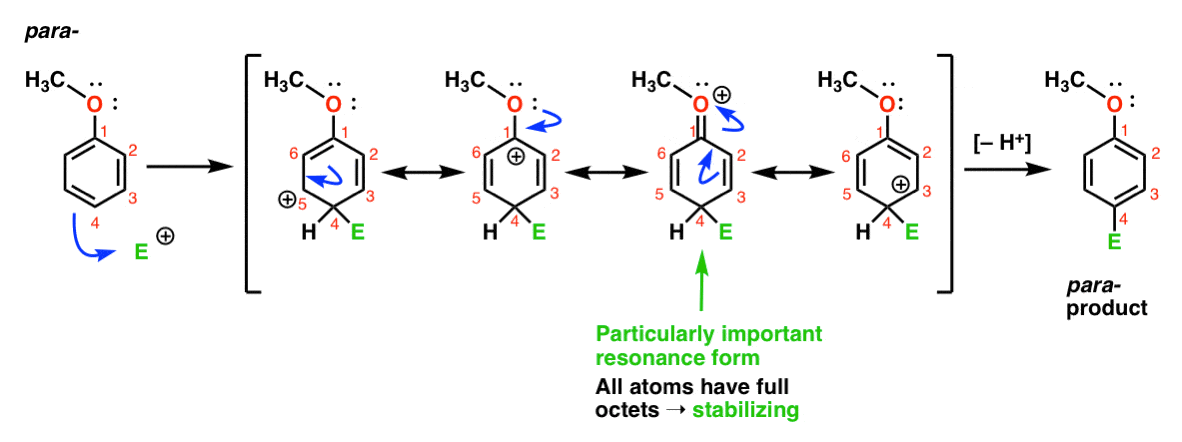
ici, nous pouvons à nouveau dessiner des formes de résonance avec des carbocations sur C1, C3 et C5, ainsi qu’une quatrième forme de résonance où l’atome d’oxygène attaché donne une paire C’est une situation essentiellement la même que l’ortho – intermédiaire.,
ainsi, en analysant les formes de résonance (et mark my word, vous pourriez bien être invité à faire de même sur un test dans un avenir proche), nous pouvons voir que les carbocations intermédiaires résultant de l’ortho – et de la para– addition sont considérablement plus stables que l’intermédiaire de la méta– addition.
ceci explique pourquoi les ortho – et para – produits dominent, et le méta – produit est mineur.
Il pourrait être utile de visualiser cela en dessinant un diagramme d’énergie de réaction.
4., Le diagramme de réaction-énergie pour un directeur Ortho Para
l’état de transition menant aux produits d’addition ortho – et para est beaucoup plus faible en énergie que le méta -.
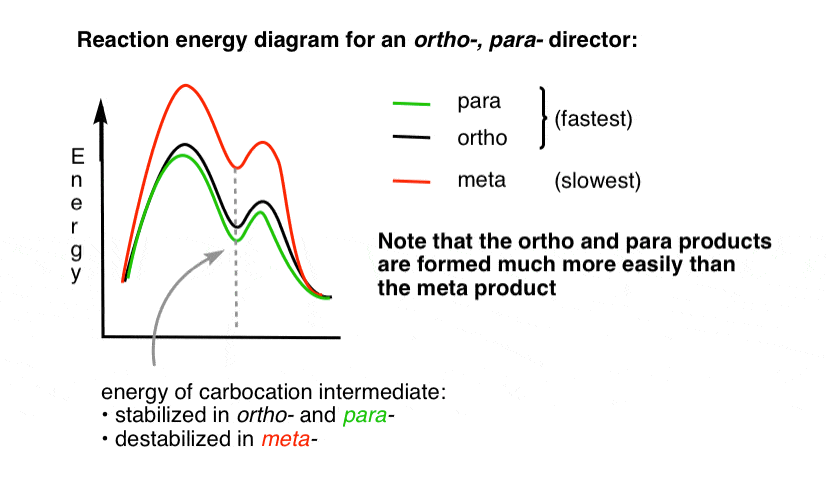
Une question – Pourquoi pensez – vous que para– pourrait être favorisé (par exemple 60% -70% de rendement pour le para-produit de nitration, ci – dessus, contre 30-40% pour ortho -) même s’il y a deux positions ortho et un seul para -?
réfléchissez à cela pendant une minute, et nous l’aborderons ci-dessous.
5. Étude de CAS No 2: un méta-directeur (CF3).
alors QU’en est-il du CF3?, Pourquoi donne-t-il méta – produits? Nous allons faire le même type d’analyse.
Voici la voie menant à l’ortho – produit. Comme pour l’ortho – intermédiaire ci-dessus, nous pouvons dessiner trois formes de résonance plaçant le carbocation sur C1, C3 et C5, respectivement.
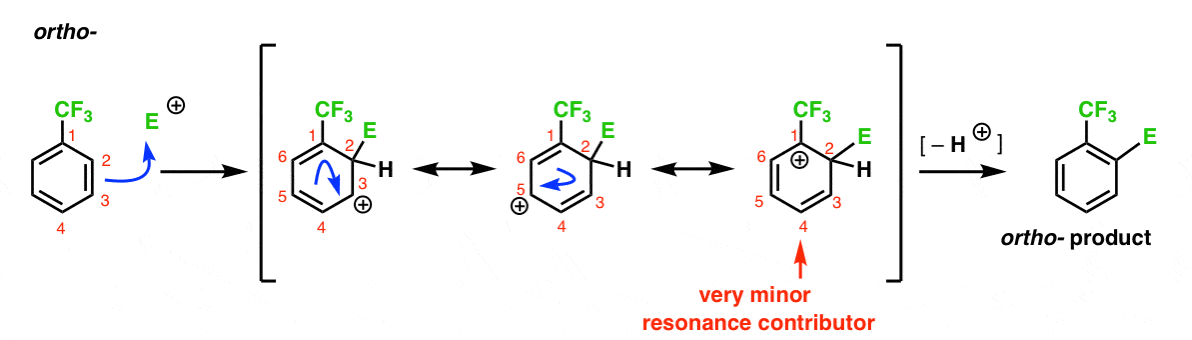
Donc ce qui est différent dans ce cas?
ce qui est différent, c’est la présence d’un fort groupe de retrait d’électrons (CF3) en C1, ce qui change complètement le jeu de balle.
Comme nous l’avons dit, les carbocations sont déstabilisées par des voisins qui retirent la densité électronique., Par conséquent, nous nous attendons à ce que ce soit un contributeur de résonance très mineur, avec le résultat que la charge positive est seulement délocalisée sur C3 et C5.
nous avons noté que CF3 est un méta – directeur. Il est raisonnable de penser qu’il y a quelque chose à propos de la méta– qui la rend particulièrement stable. En analysant l’intermédiaire pour le méta– produit ci-dessous, pouvez-vous voir pourquoi?
Hmmm. Il n’y a pas d’intermédiaire où un carbone a un octet complet. Il n’y a pas de formes de résonance particulièrement stables.
c’est un peu « meh”, n’est-ce pas?
6., Ce que nous appelons « méta– directeurs” ressemble plus à des « ortho-, para– Avoiders”
en revanche, il n’y a pas non plus de formes de résonance particulièrement instables. Étant donné que la charge positive est localisée sur C2, C4 et C6, une situation où la charge positive est directement adjacente au groupe CF3 de retrait d’électrons est évitée. Et la charge positive est ainsi bien délocalisée dans tout l’anneau, contrairement à la situation ortho, ci – dessus.
C’est donc un intermédiaire « moins mauvais” que l’ortho-.,
Enfin, le para – intermédiaire a le même problème que l’ortho- ; un intermédiaire avec une charge positive, C1, adjacente à la CF3.
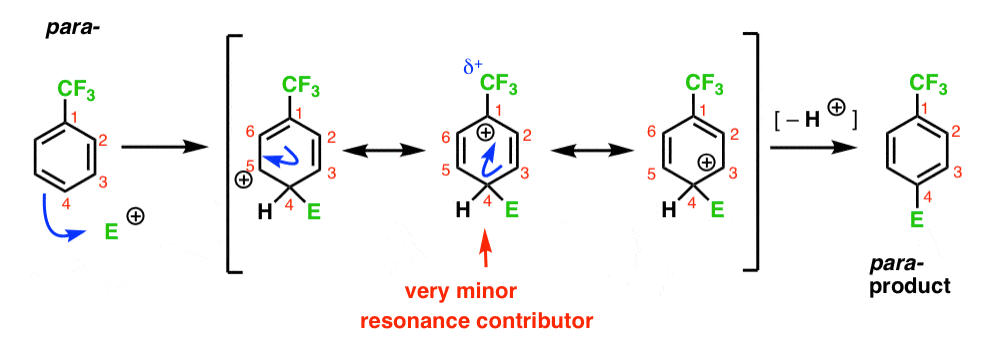
il en résulte seulement deux formes de résonance importantes, ce qui conduit à un intermédiaire de carbocation moins délocalisé (et donc moins stable).
Bottom line: l’intermédiaire méta-carbocation est plus stable que l’ortho– ou les para – intermédiaires.
Mais ce n’est pas parce que le substituant lui– même a un effet stabilisant sur le méta-intermédiaire. .,
je dirais qu’il est plus utile de considérer le méta – comme moins instable que l’ortho – et le para– .
de cette façon, ce n’est pas tant que CF3 est un méta – réalisateur, tellement qu’il est un « ortho– , para– avoider. «
7. Le diagramme D’énergie de réaction pour un méta-Directeur
Voici une esquisse du diagramme d’énergie de réaction:
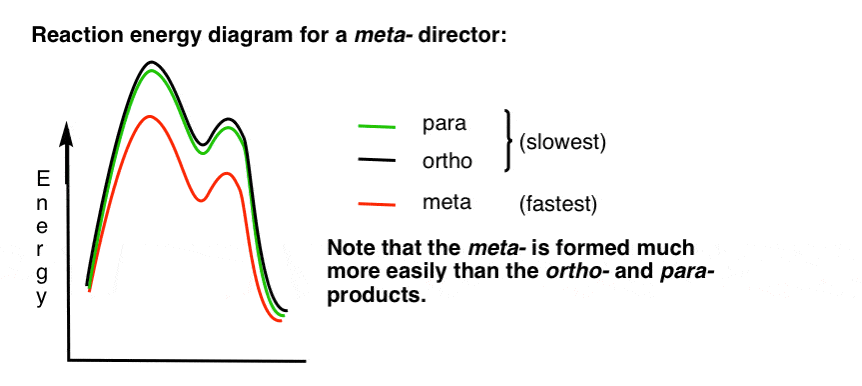
très bien.
retour à notre question initiale. Alors, quel carbocation est le plus stable?,
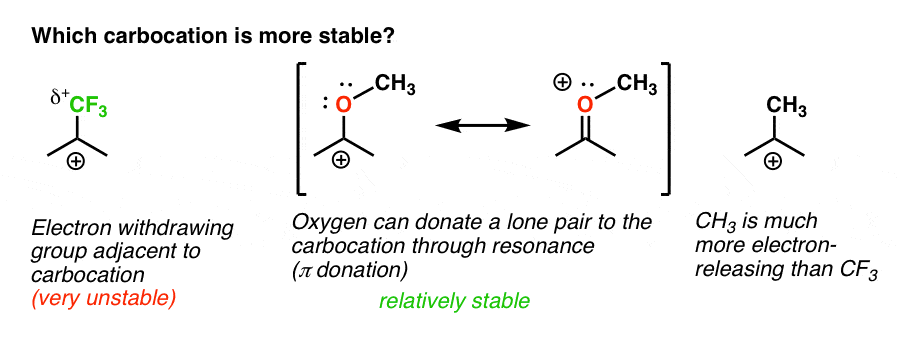
de toute évidence, le carbocation avec un oxygène adjacent portant une paire Solitaire est beaucoup plus stable qu’un carbocation adjacent à un groupe de retrait d’électrons comme CF3.
et cela comprend la différence entre un ortho-, para – directeur comme OCH3 et un méta – directeur comme CF3.
Il convient de noter que la plupart des groupes alkyles (tels que CH3 ) n’ont pas de paire isolée, mais sont toujours ortho-, para – directeurs., Ceci est cohérent avec tout ce que nous avons appris auparavant sur la façon dont les groupes alkyles se stabilisent généralement pour les carbocations – rappelez-vous que la stabilité du carbocation augmente généralement avec la substitution
donc à propos de ces para– produits
pourquoi les para – produits sont – ils produits à un taux plus élevé que l’ortho -?
la réponse est l’obstacle stérique.
L’attaque en position para est moins encombrée par le substituant que l’attaque en position ortho.
alors pourquoi les halogènes sont-ils Ortho -, Para-directeurs?
cela nous laisse avec l’exemple un peu délicat des halogènes.,
pourquoi le fluor, le chlore, le brome et l’iode sont-ils ortho -, para-directeurs même s’ils désactivent des groupes?
la réponse ne devrait pas surprendre si vous avez suivi, mais nous allons laisser cela jusqu’au prochain post, car celui-ci est déjà assez long.
Next post: pourquoi les halogènes sont-ils ortho -, para-directeurs?
Notes
Note 1. Il est plus correct de dire que les ortho – et para – produits dominent parce que les états de transition menant à ces produits sont plus faibles en énergie, plutôt que les énergies des intermédiaires eux-mêmes., Après tout, c’est l’énergie des états de transition qui détermine la barrière d’activation, et donc la vitesse de réaction.
Note 2. Ou l’hyperconjugation, que la plupart des manuels (à l’exception notable de Maitland Jones) évitent généralement.
testez-Vous!,/div>
 Cliquez pour Flip
Cliquez pour Flip
Cliquez pour Flip  Cliquez pour Flip
Cliquez pour Flip 
(Avancé) Références et lectures complémentaires
- A., F. Holleman, Die direkte Einführung von Substituenten in den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/récupérable.19100291205
A. F Holleman de 1910 a déclaré que l’orientation ortho–para est associée à l’activation et la méta-orientation à la désactivation. - —la nature de l’effet alternatif dans les chaînes carbonées. Partie XXIII. orientation anormale par les halogènes, et son incidence sur le problème du rapport ortho–para, dans la substitution aromatique
Christopher Kelk Ingold et Charles Cyril Norrey Vass
J. Chem. Soc. 1928,417-425
DOI: 10.,1039 / JR9280000417
Cet article traite des effets de direction dans les 1,2-dihalobenzènes. - quelques observations concernant l’obstacle stérique et les effets des substituants sur le rapport ortho : para dans la substitution aromatique
P. B. D. de la Mare
J. Chem. Soc. 1949, 2871-2874
DOI: 10.1039/JR9490002871
dans cet article, de la Mare discute de divers facteurs qui peuvent expliquer une prédominance du para sur l’ortho-sélectivité, après avoir divisé le rendement du produit ortho par 2 (puisqu’il y a 2 positions ortho mais seulement 1 Position para dans les benzènes monosubstitués)., - les effets de Volume des groupes alkyle en composés aromatiques. Partie V. La monosulfonation du p-cymène
R. J. W. Le Fèvre
J. Chem. Soc. 1934, 1501-1502
DOI: 10.1039/JR9340001501
Dans Le p-cymène, le produit principal obtenu par sulfonation électrophile est le produit 2 (ortho au groupe méthyle), probablement dû aux stériques. - Effects of Alkyl Groups in Electrophilic Additions and Substitutions
COHN, h., HUGHES, E., JONES, M. and PEELING, M. G.
Nature 1952, 169, 291
DOI: 1038 / 169291a0
Cet article contient des données comparant la nitration du t-butylbenzène et du toluène., Le T-butylbenzène est beaucoup plus dirigeant vers le P que le toluène (79,5% de para pour le T-butylbenzène contre 40% de para pour le toluène), ce qui est probablement dû aux sterics (l’approche ortho est bloquée par le Groupe t-butyle plus volumineux). - Distribution des isomères dans la Mononitration de L’éthyl – et de L’Isopropylbenzène. Autres preuves d’un effet stérique dans la Distribution des isomères
Herbert C. Brown et W. Hallam Bonner
Journal of the American Chemical Society 1954, 76 (2), 605-606
DOI: 10.,1021 / ja01631a084
le tableau II de cet article montre que le produit ortho obtenu à partir de la nitration des monoalkylbenzènes diminue à mesure que le groupe alkyle grossit (par exemple, le T-butylbenzène donne très peu de produit ortho lors de la nitration par rapport au toluène). - —la nature de l’effet alternatif dans les chaînes carbonées. Partie XXII. une tentative plus poussée de définir le mécanisme probable de l’orientation dans la substitution aromatique
Christopher Kelk Ingold et Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926
DOI: 10.,1039 / JR9270002918
Un premier article de L’influent physicien chimiste organique, Prof. C. K. Ingold, indiquant que les halogénobenzènes sont inductivement électron-retrait mais simultanément résonance-stabilisation. - La réactivité anormale du Fluorobenzène dans la Substitution électrophile aromatique et les phénomènes connexes
Joel Rosenthal et David I., Schuster
Journal of Chemical Education 2003, 80 (6), 679
Doi: 1021/ed080p679
Un article très intéressant, adapté aux étudiants de premier cycle curieux, et discute de quelque chose que la plupart des chimistes organiques pratiquants sauront empiriquement – le fluorobenzène est presque aussi réactif que le benzène dans les réactions EAS ou Friedel-Crafts, ce qui est contre-intuitif quand on considère les effets électroniques! - chimie organique
N. Haworth, C. K. Ingold, T. A. Henry
Ann. Rép. Prog. Chem. 1926, 23, 74-185
DOI: 10.1039/AR9262300074
Un premier article traitant des effets de direction dans EAS. Pp., 136 et 137 contiennent des figures qui représentent le flux d’électrons. Pg. 140 traite de la méta-direction, qui se produit pour les raisons « opposées » en tant que direction o/p par les groupes alkyles.