Introduction
la leucémie à cellules velues (HCL) est une néoplasie lymphoïde à cellules B. HCL se différencie des autres néoplasmes à cellules B en raison de ses caractéristiques morphologiques, immunophénotypiques et moléculaires. Sa principale caractéristique est l’accumulation de cellules B monoclonales, avec des projections ressemblant à des poils sur sa surface, principalement présentes dans le sang périphérique, la moelle osseuse et la rate. Elle a été décrite en 1958 par Bournoncle et coll. Cependant, ils l’ont appelé réticuloendothéliose leucémique.,1 Le terme « cellules velues” a été inventé en 1996 par Schrek et al.2 Aujourd’hui, il est classé par l’Organisation Mondiale de la santé comme lymphome non hodgkinien à cellules B.3
définition
le HCL est un trouble lymphoprolifératif chronique rare des cellules B. Ses principales caractéristiques sont les prolongements cytoplasmiques qui donnent aux cellules un aspect Poilu.
étiologie
bien qu’il existe différentes études montrant un lien entre l’exposition à certains agents et le HCL, son étiologie n’a pas été clairement établie, ce qui pourrait s’expliquer par sa faible incidence., Parmi les agents qui ont prouvé un lien positif figurent: exposition aux pesticides, 4 herbicides, huiles minérales, travail de charpentier ou d’agriculteur.4-7 récemment, un lien positif avec la taille a été décrit,5 alors que le tabagisme a une association inverse, en particulier chez les hommes.8 le mécanisme spécifique qui confère la protection est inconnu; néanmoins, le tabagisme a été suggéré pour diminuer la sévérité des mécanismes inflammatoires9 et que la nicotine induit l’apoptose dans les lymphocytes.10
épidémiologie
Les HCL représentent 2 à 3% de toutes les leucémies. Il y a 600 nouveaux cas signalés par an aux États-Unis (3.,2 cas par million d’habitants). L’âge médian au diagnostic est de 52 ans et il est plus fréquent chez les hommes que chez les femmes, avec un rapport de 4:1; avec une incidence plus élevée dans la population blanche, en particulier chez les Juifs ashkénazes.3 au Mexique, le HCL représente 1,12% de toutes les leucémies. Cependant, dans le nord du pays,il représente jusqu’à 1,83%, similaire à l’information des États-Unis 11, 12
physiopathologie
le HCL est un trouble lymphoprolifératif chronique à cellules B. Cependant, ses cellules n’ont pas l’apparence d’une sous-population de cellules B, et son origine a fait l’objet de débats., L’analyse des gènes des régions variables des immunoglobulines (Ig) est un outil utilisé pour découvrir l’origine clonale des cellules lymphoïdes.13 dans plus de 85% des cas, nous sommes en mesure de trouver des mutations somatiques dans les gènes des régions variables Ig des cellules HCL,14,15, ce qui indique que les cellules impliquées ont traversé le centre germinal ou les organes périphériques lymphoïdes.16 environ 40% des cellules HCL Co-expriment de multiples isotypes D’Ig liés au clonage.17
Les preuves suggèrent une origine post-centre germinal dans les cellules B de la mémoire, en raison de leur profil d’expression génomique.,18,19 L’origine des cellules B de la mémoire est compatible avec L’absence de translocation réciproque chromosomique de HCL.20 L’Absence de CD27 est typique en HCL. Cela représente un point contre l’hypothèse de son origine dans les cellules B de mémoire.21 cependant, des cellules B à mémoire CD27 négatives ont été observées.22 Comme l’affection des ganglions lymphatiques est peu fréquente, il a été suggéré que la cellule D’origine du HCL est probablement située dans la moelle osseuse ou la rate, car ce sont généralement les endroits les plus touchés. Les cellules HCL présentent un profil d’expression similaire à la zone marginale de la rate.,23
L’aspect caractéristique du HCL est dû à l’expression de la bêta-actine, qui est polymérisée en F-actine, située dans le cytosquelette cortical.La phosphoprotéine 24 PP52, spécifique des leucocytes, est liée à la F-actine et est responsable du soutien des projections ressemblant à des cheveux.25 d’autre part, le séquençage du gène HCL a récemment identifié la présence d’une mutation BRAF V600E chez presque tous les patients atteints de la maladie, absente dans d’autres tumeurs malignes lymphoïdes à cellules B.26,27 mutations BRAF activent la voie MAPK, favorisant la croissance, la survie et la différenciation cellulaire HCL.,28
présentation clinique et résultats de laboratoire
l’évolution clinique de la maladie est indolente. La plupart des patients présentent généralement de la faiblesse et de la fatigue comme symptômes prédominants au début de la maladie.29 Parfois, il y a des antécédents d’infections répétées. Les résultats de l’examen fisical sont: splénomégalie dans 96%, hépatomégalie dans 58% et lymphadénopathie dans seulement 35%. Ces ganglions lymphatiques enflés sont rarement observés en périphérie; néanmoins, ils sont généralement présents dans l’abdomen et détectés par des études d’imagerie.,30
à un stade avancé de la maladie, nous pouvons trouver des douleurs dans le quadrant supérieur gauche, des infections, de la fièvre, des hémorragies et / ou une perte de poids. Cependant, cela est rare en raison de la disponibilité et de l’efficacité du traitement.30,31 les manifestations cliniques sont le produit d’une accumulation de cellules velues dans la rate, le foie et la moelle osseuse (Tableau 1).32
les manifestations Cliniques.,
| Rate | Foie | de la moelle Osseuse | ganglions Lymphatiques |
|---|---|---|---|
| Les cellules s’accumulent dans la pulpe rouge et l’atrophie de la pulpe blanche. Ils forment plus tard les soi-disant” pseudo-sinusoïdes » par le remplacement des cellules endothéliales de la pulpe rouge par des canaux vasculaires améliorés, contribuant à l’anémie. | Ici, ils s’accumulent dans les sinusoïdes hépatiques ainsi que dans le portail des voies., Il y a une fibrose dans ce dernier en raison de l’acide hyaluronique abondant, qui stimule les cellules velues à produire de la fibronectine, avec sa fibrose correspondante. | dans ce domaine, il existe une production extensive de fibrose et une suppression de l’hématopoïèse. Le facteur clé est l’interaction des cellules velues avec l’acide hyaluronique de la matrice extracellulaire, générant des facteurs de croissance fibroblastiques (FGF) et stimulant les cellules malignes pour produire et sécréter de la fibronectine. | généralement, absent de la maladie. Manque de récepteurs pour l’entrée des cellules velues., |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosis
HCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v)., Le dernier survient dans 10% des cas avec un âge médian de 70 ans, et malgré les similitudes avec la leucémie à cellules velues classique, ils divergent en l’absence des marqueurs immunophénotypiques CD25 et CD123. Une autre façon de faire un diagnostic différentiel est l’absence de réponse au traitement HCL standard et l’inexistence de mutations du gène BRAF V600F.30
méthodes de Diagnostic
le diagnostic de HCL est généralement effectué par biopsie et aspiration de moelle osseuse combinée à une caractérisation immunophénotypique par cytométrie en flux.,33 Il est important de souligner que cette pathologie est généralement sous-diagnostiquée et nécessite une suspicion clinique et de l’utilisation de la technologie pour résoudre ce problème. Comme mentionné précédemment, la plupart des patients (70-90%) présentent une pancytopénie, une leucopénie (
×109/L), une anémie (g/dL), une neutropénie (×109/L), une monocytopénie (×109/L) et une thrombocytopénie (×109/L). Seulement entre 10% et 20% présentent une leucocytose modérée (>10×109/L). Les patients atteints de HCL présentent des taux sériques élevés D’IL-2R (CD25), qui sont en corrélation avec le degré d’activité de la maladie.,34 les autres tests à prendre en compte lors du diagnostic sont les taux sériques d’immunoglobulines, ainsi que les mutations somatiques du gène IgVH et du BRAF V600E.32 certaines caractéristiques histopathologiques et immunophénotypiques du HCL sont les suivantes:
- •
cytométrie en flux lymphocytaire dans le sang périphérique ou la moelle osseuse avec des expressions CD19, CD20, FMC7, CD11c, CD103, CD25, HC2, CD22, sIg, CD79a et CD123, quatre étant les marqueurs principaux et spécifiques: CD11c, CD103, CD25 et CD123.CD5, CD23, CD10, cd79b et CD27.,32
- •
une forte expression de CD200 est caractéristique de HCL et peut être utile dans le diagnostic de cas difficiles.34
- •
aspiration de la moelle Osseuse avec une aiguille peut être difficile à obtenir et souvent improductive ou « sec”. Dans une biopsie de moelle osseuse, nous pouvons observer une fibrose, avec un aspect cellulaire « œuf frit » causé par les grands espaces entre les noyaux et le cytoplasme abondant. Des analyses immunohistochimiques de la CD20 et de la TRAP (phosphatase acide résistante au tartrate), de la DBA-4 et de l’annexine A1 sont effectuées, qui sont caractéristiquement positives.,32
Andrulis et coll. dirigé une étude où l’efficacité de L’anticorps VE1 pour la détection de BRAF V600E a été rapportée, ainsi que l’identification de HCL dans d’autres entités. De plus, une étude menée par Uppal et al. trouvé une sensibilité de 88% et une spécificité de 97% pour détecter cette mutation à l’anticorps.34
traitement actuel
le HCL a régulièrement une évolution indolente. L’attente et l’observation sont une bonne option pour les patients asymptomatiques, car un traitement précoce n’offre aucune sorte d’avantage dans les taux de survie de ces cas., Quoi qu’il en soit, la progression de la maladie chez la plupart des patients entraînera des complications à la suite de cytopénies et de splénomégalie; c’est-à-dire anémie, hémorragies, infections récurrentes, etc. En pratique générale, le traitement doit être commencé si l’un des critères énumérés dans le tableau 2 se produit.35 lorsque la décision de ne pas commencer le traitement est prise, une surveillance clinique et de laboratoire doit être effectuée tous les trois mois pendant la première année et tous les six mois par la suite.,35 le traitement par HCL n’est pas considéré comme une guérison; mais les stratégies de traitement actuelles sont capables d’atteindre des rémissions prolongées, augmentant ainsi les taux de survie mondiaux (algorithme 1).36-39
Critères pour commencer un traitement.
1. Splénomégalie symptomatique
2. Cytopénie impliquant au moins un des éléments suivants:
-de l’Hémoglobine g/dL
-Plaquettes ×109/L
-Neutrophiles ×109/L
3., Des infections graves,
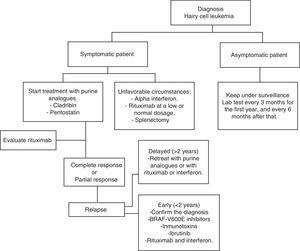
tableau pour la prise en charge d’un patient atteint de leucémie à cellules velues.
analogues des purines
en 1960, les résultats ont montré que 30% des enfants atteints du syndrome d’immunodéficience combinée sévère n’avaient pas d’enzyme désaminase adénosine (ADA).,40 de plus, ils ont découvert que l’accumulation de la forme désoxyadénosine triphosphate était responsable de la diminution des lymphocytes.41 en gardant ces observations à l’esprit, des médicaments capables de se lier de manière irréversible à L’ADA ou d’en antagoniser l’action ont été développés. Après traitement avec des analogues de purine, l’accumulation de désoxyadénosine triphosphate entraîne une rupture et une inhibition de la réparation de l’ADN, ce qui se traduit par une apoptose cellulaire.,42
Pentostatine
également connue sous le nom de 2′-désoxycoformycine (dcF), un produit de Streptomyces antibioticus et d’inhibiteurs de L’ADA, introduit pour la première fois en 1980 en tant que premiers analogues de purine pour le traitement du HCL. Dans le tableau 3,37,39,43,44 des études spécifiques montrent des taux de réponse à la pentostatine. Utilisation intraveineuse de pentostatine à un rapport de 4 mg/m2 une fois toutes les deux semaines jusqu’à ce que la réponse complète soit approuvée aux États-Unis.La Pentostatine est sans danger chez les patients présentant un éclaircissement de la créatinine >60 ml/min. Cependant, une réduction de dosage est nécessaire si cette dépuration est comprise entre 40 et 60ml/min.,32 l’hydratation du Patient avec 1,5 L de solution intraveineuse à chaque cycle de pentostatine est recommandée.30
la réponse globale varie entre 88% et 96%, tandis que la réponse complète se situe entre 44% et 81%. Flinn et coll. estimation d’un taux de survie de 5 et 10 ans de 90% et 81%, respectivement, avec une durée moyenne de suivi de 9,3 ans.45 La Pentostatine est généralement bien tolérée et les effets indésirables les plus courants sont l’anémie, la thrombocytopénie et la neutropénie.,46 pourtant, on a rapporté que la pentostatine diminuait significativement le nombre de lymphocytes CD4+ et CD8+, ce qui pourrait augmenter les tumeurs malignes secondaires et l’incidence des infections.47,48
Cladribine
il est connu sous le nom de 2-chlorodésoxyadénosine (CdA). Dans le tableau 4,49-52, les études indiquent son efficacité. Le schéma le plus utilisé consiste à administrer 0,1 mg/kg/jour en perfusion continue pendant 7 jours. Dans une étude non randomisée, il y avait des preuves prouvant qu’il n’y avait pas de différence statistiquement significative dans les gammes de réponse et de toxicité entre les perfusions (24h et 2h).,53 Une autre étude randomisée a comparé l’administration quotidienne par rapport à l’administration hebdomadaire de cladribine; il n’y a pas eu de résultats significatifs en termes de réponse, de survie, de taux global et de toxicité.54 une autre étude a montré que le programme hebdomadaire réduisait le risque d’infections.55 Un des avantages de l’administration sous-cutanée est le fait que dans la plupart des cas, il ne nécessite pas d’hospitalisation. 0,14 mg / jour pendant 5 jours a un taux de réponse de 95%, 56 similaire à l’administration intraveineuse. Les programmes sous-cutanés hebdomadaires ont des taux de réponse et de toxicité similaires à ceux quotidiens.,57 avec un seul cycle de cladribine, une réponse globale pouvant atteindre 100% peut être obtenue, et les taux de réponse totaux diffèrent de 77% à 95%.49-52 Jehn et coll. rapporté une survie globale à 12 ans est de 79%.36 en général, la cladribine est bien tolérée, les cytopénies et la fièvre étant les effets indésirables les plus fréquents.
études sur l’efficacité de la cladribine dans les cas de HCL.,
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days |
77.3 | 18.,6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days |
88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days |
95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.,1mg/kg/d 7 days |
79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence., Malgré cela, il existe des études rétrospectives prouvant le fait que les deux médicaments ont une efficacité similaire, en termes de réponse complète et de survie sans maladie.
autres traitements
les analogues des purines restent la première ligne de traitement mais de nouvelles découvertes concernant la physiopathologie des HCL ont conduit à la création de médicaments avec différentes cibles thérapeutiques. Ces médicaments sont en cours de recherche et certains ont montré des résultats prometteurs.
Rituximab
puisque le HCL est une tumeur maligne à cellules B, il est logique d’utiliser un anticorps monoclonal contre le CD20, tel que le rituximab., Utilisé comme médicament autonome, le rituximab peut atteindre des taux de réponse totale de 10 à 54% chez les patients présentant une rechute de HCL, à une dose de 375 mg/m2 une fois par semaine pendant 4 à 8 semaines.58,59 d’Autre et coll. a examiné rétrospectivement 18 patients traités par des analogues de la purine en association avec le rituximab en deuxième ligne de traitement, après avoir été traités par des analogues de la purine en monothérapie. Tous les patients ont répondu, avec un taux de réponse complet de 89%.60
Rituximab à 375mg / m2 par semaine pendant 8 semaines comme traitement initial après administration de 5.,6 mg/m2 de cladribine par perfusion IV à 2 h pendant 5 jours génère une réponse totale de 100%.61 dans des situations spéciales ou défavorables, 100 mg de rituximab par semaine peuvent être utilisés pendant 4 à 6 semaines. Ceci est moins coûteux et est généralement efficace, surtout lorsqu’il est combiné avec de l’interféron.
bien que les analogues des purines ne puissent pas éliminer le HCL, étant donné que la maladie résiduelle minimale (MRD) détectée après l’administration de cladribine est toujours fortement CD20+, l’éradication de MRD peut être obtenue en utilisant le rituximab. Rivandi et coll., prouvé dans une étude préliminaire que le rituximab à des doses conventionnelles pendant une période de 8 semaines fonctionne avec une grande activité, éliminant la MRD chez 13 patients, quand il est utilisé 4 semaines après l’administration de cladribine.62
Vemurafenib
comme décrit précédemment, la mutation BRAF V600E est la clé génétique du HCL. C’est donc une cible thérapeutique qui a été étudiée ces dernières années. Vemurafenib est un inhibiteur oral de BRAF V600E. Tiacci et coll., a mené une étude pour mesurer L’activité et la sécurité du Vemurafenib chez des patients atteints de HLC qui ont rechuté après un traitement par des analogues de purine ou qui étaient réfractaires à l’administration d’analogues de purine. Le taux de réponse Global était de 96% et le taux de réponse complète de 35%, avec une moyenne de survie sans rechute de 19 mois. Les effets indésirables ont été des éruptions cutanées, des arthralgies et de l’arthrite.63
comme une relation positive a été observée entre L’utilisation de Vemurafenib et l’apparition de tumeurs malignes dermatologiques, des explorations fréquentes de la peau sont recommandées.,
Ibrutinib
un inhibiteur sélectif et irréversible de la tyrosine kinase de Burton intervient dans la voie de signalisation des cellules B.64 un essai clinique d’ibrutinib chez les patients présentant une rechute de HCL a récemment commencé. Les données préliminaires sur l’efficacité et l’innocuité montrent des effets indésirables tels que des éruptions cutanées, de la diarrhée et de l’arthralgie. Ce test clinique est actuellement en cours dans plusieurs centres aux États-Unis (NCT01841723).
immunotoxines
afin d’augmenter la cytotoxicité des anticorps monoclonaux, des techniques facilitant la production de conjugués anticorps–toxine ou anticorps–médicament ont été créées., Une immunotoxine est la fusion entre une toxine bactérienne (exotoxine de Pseudomonas ou diphtérie) et la fraction variable d’un anticorps monoclonal, dont la cible spécifique se trouve à la surface des cellules néoplasiques comme CD25 ou CD22. Cette toxine est libérée dans l’intérieur de la cellule néoplasique et interfère avec la synthèse des protéines.65
BL22 est une immunotoxine contre CD22 fusionnée avec une forme tronquée de L’exotoxine P. PE38. Dans un essai clinique de phase II, BL22 a été testé dans 36 cas de rechute de HCL ou de maladie réfractaire., Après un cycle (40mg / kg tous les deux jours, trois doses), le taux de réponse complet était de 25% et le taux de réponse global était de 50%. Ces réponses se sont améliorées pour atteindre un taux de réponse total de 47% et un taux de réponse global de 72% après retraitement (uniquement chez les patients atteints de cytopénies). Deux patients ont développé un syndrome hémolytique urémique sans qu’il soit nécessaire de recourir à la plasmaphérèse.66 par la suite, moxetumomab pasudotox a été développé comme une version modifiée de BL22 avec une affinité et une cytotoxicité plus élevées., Dans un essai de phase I, qui comprenait 28 patients présentant une rechute et une résistance au HCL, un taux de réponse global de 86% a été obtenu, y compris un taux de réponse complète soutenue chez 46% des patients.67
options thérapeutiques dans des circonstances défavorables
même si le HCL est traité dans la plupart des pays développés avec de la cladribine et de la pentostatine, il est un fait que ces médicaments ne sont pas seulement coûteux, ils ne sont pas disponibles au Mexique et dans de nombreux pays aux ressources limitées., Ainsi, dans ces types de circonstances, il existe d’autres options thérapeutiques abordables avec des résultats favorables
L’interféron alpha pour le traitement des patients HCL a été introduit pour la première fois en 1984. Aujourd’hui, son utilisation est limitée, principalement en raison de la grande efficacité des analogues des purines. D’autre part, dans les pays à faibles ressources économiques, il s’agit d’une option peu coûteuse et qui a donné des résultats similaires à ceux de la cladribine en termes de survie mondiale. Ruiz-Delgado et coll., a mené une étude comparative entre l’interféron alpha (n=18) et la cladribine (n=11), où la différence de survie globale entre les deux groupes n’était pas statistiquement significative; 94% à 217 mois dans le groupe interféron et 91% à 133 mois dans le groupe cladribine.68 dans une étude menée dans notre centre, neuf patients HCL ont reçu trois méga-unités IFN trois fois par semaine pendant 12 semaines, puis ils ont reçu un traitement à nouveau pendant 8 semaines lorsqu’il y avait une réactivation de la leucémie ou après 10 mois d’observation chaque année., Tous les patients ont eu une rémission hématologique avant 12 semaines de traitement. Cette option thérapeutique est moins chère, efficace et comparable à d’autres formes de thérapie avec IFN dans le traitement et le maintien des patients atteints de ce type de leucémie.69 Il est possible d’associer l’interféron au rituximab sans augmenter les effets toxiques et améliorer l’efficacité.
la splénectomie a été la première intervention qui a modifié significativement la survie chez les patients. Aujourd’hui, il est rarement utilisé., Il peut être recommandé chez les patients présentant une splénomégalie massive douloureuse (> 10 cm sous le bord costal) et une infiltration minimale de la moelle osseuse, ou chez les patients réfractaires au traitement par interféron et analogues de la purine.33 études rétrospectives montrent un taux de réponse complet de 40 à 62% et un taux de survie moyen à 5 ans jusqu’à 68%.70,71 Garçon et coll. publié une étude rétrospective, y compris 24 patients avec un diagnostic de HCL, ils ont été divisés en deux groupes: 17 patients ont reçu de la cladribine et 7 ont été splénectomisés., 75% des patients du groupe splénectomie ont montré une rémission totale, 94% l’ont fait dans le groupe cladribine. Une conclusion intéressante en comparant les deux groupes était qu’aucune différence statistiquement significative n’a été observée concernant la survie sans leucémie et la survie globale.72
pronostic
le temps de survie des patients après le diagnostic était de 4 ans avant qu’un traitement ne soit connu, en raison de complications dérivées des cytopénies, y compris des hémorragies et des infections. Par la suite, avec la splénectomie comme traitement de première intention, il y a eu une réponse complète de 40-62% et un taux de survie de 5 ans à 61-68%., Ensuite, l’interféron alfa a été utilisé comme premier médicament présentant des avantages dans le traitement de L’HCL. Pourtant, son taux de réponse complet était faible, à 10%.73
aujourd’hui, avec les analogues de la purine (pentostatine et cladribine), une réponse complète est induite jusqu’à 80% des patients avec une survie médiane de 10 ans. Les taux de réponse globaux sont 96-100% avec un taux de réponse de 80% et un taux de survie de 10 ans comprise entre 85% et 100%.74 malgré cela, une proportion importante de patients atteints de HCL échouent dans leur réponse au traitement ou deviennent résistants., Jusqu’à 48% des patients rechutent au cours des 15 années suivantes.75 l’avenir des patients HCL est très favorable. Le défi consiste à identifier cette malignité le plus tôt possible et à la traiter correctement en utilisant les ressources disponibles.
Conflit d’intérêts
Les auteurs n’ont aucun conflit d’intérêts à déclarer.