Einleitung
Hairy cell leukemia (HCL) ist ein B-Zell-lymphoiden Neoplasien. HCL unterscheidet sich aufgrund seiner morphologischen, immunphänotypischen und molekularen Eigenschaften von anderen B-Zell-Neoplasmen. Sein Hauptmerkmal ist die Ansammlung monoklonaler B-Zellen mit haarartigen Projektionen auf ihrer Oberfläche, die hauptsächlich im peripheren Blut, im Knochenmark und in der Milz vorkommen. Es wurde 1958 von Bournoncle et al. Sie nannten es jedoch leukämische Retikuloendotheliose.,1 Der Begriff „haarige Zellen“ wurde 1996 von Schrek et al.geprägt.2 Heute wird es von der Weltgesundheitsorganisation als B-Zell-Non-Hodgkin-Lymphom eingestuft.3
Definition
HCL ist eine seltene, chronische, lymphoproliferative B-Zellen-Störung. Seine Hauptmerkmale sind die zytoplasmatischen Verlängerungen, die den Zellen einen haarigen Aspekt verleihen.
Ätiologie
Obwohl es verschiedene Studien gibt, die einen Zusammenhang zwischen der Exposition gegenüber bestimmten Wirkstoffen und HCL zeigen, wurde ihre Ätiologie nicht eindeutig festgestellt, was aufgrund ihrer geringen Inzidenz erklärt werden könnte., Zu den Wirkstoffen, die sich als positiv erwiesen haben,gehören: Exposition gegenüber Pestiziden, 4 Herbiziden, Mineralölen, Arbeiten als Tischler oder Landwirt.In jüngster Zeit wurde ein positiver Zusammenhang mit der Größe beschrieben, 5 während Rauchen insbesondere bei Männern eine umgekehrte Assoziation aufweist.8 Der spezifische Mechanismus, der den Schutz verleiht, ist unbekannt; dennoch wurde das Rauchen vorgeschlagen, um die Schwere der Entzündungsmechanismus9 zu verringern und dass Nikotin Apoptose in Lymphozyten induziert.10
Epidemiologie
HCL macht 2-3% aller Leukämien aus. Es gibt 600 neue Fälle pro Jahr in den USA gemeldet (3.,2 Fälle pro million Einwohner). Das Durchschnittsalter bei der Diagnose ist 52 Jahre alt und bei Männern häufiger als bei Frauen mit einem Verhältnis von 4:1; mit einer höheren Inzidenz in der weißen Bevölkerung, insbesondere bei aschkenasischen Juden.3 In Mexiko macht HCL 1, 12% aller Leukämien aus. Im Norden des Landes sind es jedoch bis zu 1,83%, ähnlich den Informationen aus den USA 11, 12
Physiopathologie
HCL ist eine chronische, lymphoproliferative B-Zell-Störung. Seine Zellen haben jedoch nicht das Aussehen einer B-Zell-Unterpopulation, und sein Ursprung war umstritten., Die Analyse von Immunglobulinen (Ig) variabler Regionen Gene ist ein Werkzeug, um den klonalen Ursprung der lymphatischen Zellen zu entdecken.13 In über 85% der Fälle können wir somatische Mutationen in Ig-variablen Regionen Gene von HCL-Zellen finden, 14, 15 was ein Hinweis darauf ist, dass die beteiligten Zellen das Keimzentrum oder lymphoide periphere Organe passiert haben.16 Etwa 40% der HCL-Zellen exprimieren mehrere klonal verwandte Ig-Isotypen.17
Hinweise deuten auf einen Ursprung des Keimzentrums in Speicher-B-Zellen aufgrund ihres genomischen Expressionsprofils hin.,18,19 Der Ursprung der Speicher-B-Zellen ist mit dem Mangel an chromosomischer reziproker Translokation von HCL kompatibel.Das Fehlen von CD27 ist typisch für HCL. Dies stellt einen Punkt gegen die Hypothese seines Ursprungs in Speicher-B-Zellen dar.21 Jedoch wurden negative CD27-Speicher-B-Zellen beobachtet.22 Da eine Lymphknotenerkrankung selten vorkommt, wurde vorgeschlagen, dass sich die HCL-Ursprungszelle wahrscheinlich im Knochenmark oder in der Milz befindet, da dies normalerweise die am stärksten betroffenen Stellen sind. HCL-Zellen zeigen ein Expressionsprofil ähnlich der Randzone der Milz.,23
Das charakteristische Aussehen von HCL beruht auf der Beta-Aktin-Expression, die zu F-Aktin polymerisiert ist und sich im kortikalen Zytoskelett befindet.24 PP52 Phosphoprotein, das für Leukozyten spezifisch ist, ist mit F-Aktin verbunden und dafür verantwortlich, die haarartigen Projektionen zu unterstützen.25 Auf der anderen Seite identifizierte die HCL-Gensequenzierung kürzlich das Vorhandensein einer BRAF V600E-Mutation bei fast jedem Patienten mit der Krankheit, der bei anderen B-Zell-lymphatischen Malignomen fehlte.26,27 BRAF-Mutationen aktivieren den MAPK-Weg und fördern Wachstum, Überleben und HCL-Zelldifferenzierung.,28
Klinische Darstellung und Laborbefunde
Der klinische Verlauf der Erkrankung ist indolent. Die meisten Patienten zeigen normalerweise Schwäche und Müdigkeit als vorherrschende Symptome während des Ausbruchs der Krankheit.aber manchmal gibt es eine Geschichte von wiederholten Infektionen. Die fisical Untersuchungsergebnisse sind: Splenomegalie in 96%, Hepatomegalie in 58% und Lymphadenopathie in nur 35%. Diese geschwollenen Lymphknoten werden selten in der Peripherie beobachtet; Dennoch sind sie im Allgemeinen im Abdomen vorhanden und werden durch bildgebende Untersuchungen nachgewiesen.,30
In einem fortgeschrittenen Stadium der Krankheit können wir Schmerzen im oberen linken Quadranten, Infektionen, Fieber und Blutungen und/oder Gewichtsverlust feststellen. Dies ist jedoch aufgrund der Verfügbarkeit und Wirksamkeit der Behandlung ungewöhnlich.30,31 Klinische Manifestationen sind Produkt einer Haarzellansammlung in Milz, Leber und Knochenmark (Tabelle 1).32
Klinische Manifestationen.,
| Milz | Leber | Knochenmark | Lymphknoten |
|---|---|---|---|
| Die Zellen sammeln sich in der roten Pulpa an und verkümmern die weiße Pulpa. Sie bilden später die sogenannten „Pseudo-Sinusoide“ durch den Ersatz der Endothelzellen der roten Pulpa durch verbesserte Gefäßkanäle, die zur Anämie beitragen. | Hier sammeln sie sich sowohl in den Leber-Sinusoiden als auch im Portaltrakt an., Es gibt Fibrose in letzterem aufgrund der reichlich vorhandenen Hyaluronsäure, die die behaarten Zellen stimuliert, Fibronektin mit seiner entsprechenden Fibrose zu produzieren. | In diesem Bereich gibt es eine ausgedehnte Produktion von Fibrose und Unterdrückung der Hämatopoese. Der Schlüsselfaktor ist die Wechselwirkung der behaarten Zellen mit der Hyaluronsäure der extrazellulären Matrix, die fibroblastische Wachstumsfaktoren (FGF) erzeugt und die Malignität stimuliert, um Fibronektin zu produzieren und abzusondern. | Im Allgemeinen fehlt die Krankheit. Mangel an Rezeptoren für den Eintritt von behaarten Zellen., |
Respect to lab studies, it is frequent to observe anemia in 85%, thrombocytopenia in 60.80% and leukopenia in 60% due to hypersplenism and bone marrow infiltration.30
Differential diagnosis
HCL must be differentiated from other indolent lymphoid malignancies such as prolymphocytic leukemia, splenic marginal zone lymphoma, mantle cell lymphoma and HCL variant (HCL-v)., Die letzte tritt in 10% der Fälle mit einem Durchschnittsalter von 70 Jahren auf, und trotz der Ähnlichkeiten mit der klassischen Haarzellleukämie divergieren sie in Abwesenheit der immunphänotypischen Marker CD25 und CD123. Eine andere Möglichkeit, eine Differentialdiagnose zu stellen, ist das fehlende Ansprechen auf die Standard-HCL-Behandlung und das Nichtbestehen von Mutationen des BRAF V600F-Gens.30
Diagnostische Methoden
Die HCL-Diagnose wird üblicherweise mit einer Biopsie und Knochenmarkaspiration in Kombination mit einer immunphänotypischen Charakterisierung durch Durchflusszytometrie gestellt.,33 Es ist wichtig darauf hinzuweisen, dass diese Pathologie in der Regel nicht diagnostiziert wird und einen klinischen Verdacht und die Verwendung der richtigen Technologie zur Lösung dieses Problems erfordert. Wie bereits erwähnt, weisen die meisten Patienten (70-90%) eine Panzytopenie mit Leukopenie (
×109/L), Anämie (g/dL), Neutropenie (×109/L), Monozytopenie (×109/L) und Thrombozytopenie (×109/L) auf. Nur zwischen 10% und 20% weisen eine moderate Leukozytose auf (>10×109/L). HCL-Patienten zeigen erhöhte IL-2R (CD25) – Serumspiegel, was mit dem Aktivitätsgrad der Krankheit korreliert.,34 Weitere Tests, die bei der Diagnose berücksichtigt werden sollten, sind Serumimmunglobulinspiegel sowie somatische Mutationen des IgVH-Gens und des BRAF V600E.32 Einige histopathologische und immunphänotypische Merkmale von HCL sind folgende:
- •
Lymphozytometrie im peripheren Blut oder Knochenmark mit CD19 -, CD20 -, FMC7 -, CD11c -, CD103 -, CD25 -, HC2 -, CD22 -, sIg -, CD79a-und CD123-Ausdrücken, wobei vier die wichtigsten und spezifischen Marker sind: CD11c, CD103, CD25 und CD123.34 Häufig sind negative Marker CD5, CD23, CD10, CD79b und CD27.,32
- •
Ein starker Ausdruck für CD200 ist charakteristisch für HCL und kann bei der Diagnose schwieriger Fälle nützlich sein.34
- •
Die Knochenmarkaspiration mit einer Nadel kann schwierig sein und ist häufig unproduktiv oder“trocken“. In einer Knochenmarksbiopsie können wir Fibrose mit einem zellulären „Spiegelei“ beobachten, das durch die weiten Räume zwischen Kernen und reichlich vorhandenem Zytoplasma verursacht wird. Es werden immunhistochemische Analysen für CD20 und TRAP (tartratresistente saure Phosphatase), DBA-4 und Annexin A1 durchgeführt, die charakteristisch positiv sind.,32
Andrulis et al. leitete eine Studie, in der die Effizienz des VE1-Antikörpers für den BRAF V600E-Nachweis zusammen mit der HCL-Identifizierung in anderen Entitäten berichtet wurde. Darüber hinaus eine Studie von Uppal et al. fand eine Empfindlichkeit von 88% und eine Spezifität von 97%, um diese Mutation mit dem genannten Antikörper nachzuweisen.34
Aktuelle Behandlung
HCL hat regelmäßig eine indolente Entwicklung. Warten und Beobachten ist eine gute Option für asymptomatische Patienten, da eine frühzeitige Behandlung keinen Nutzen für die Überlebensraten dieser Fälle bietet., Wie auch immer, das Fortschreiten der Krankheit bei den meisten Patienten führt zu Komplikationen als Folge von Zytopenie und Splenomegalie; d. H. Anämie, Blutungen, wiederkehrende Infektionen usw. In der allgemeinen Praxis muss mit der Behandlung begonnen werden, wenn eines der in Tabelle 2 aufgeführten Kriterien auftritt.35 Wenn die Entscheidung getroffen wird, die Behandlung nicht zu beginnen, sollte die klinische und Laborüberwachung im ersten Jahr alle drei Monate und danach alle sechs Monate durchgeführt werden.,35 Die HCL-Behandlung gilt nicht als heilend; Aktuelle Behandlungsstrategien sind jedoch in der Lage, verlängerte Remissionen zu erreichen und so die globale Überlebensrate zu erhöhen (Algorithmus 1).36-39
Kriterien, um die Behandlung zu beginnen.
1. Symptomatische Splenomegalie
2. Zytopenie mit mindestens einem der folgenden:
– Hämoglobin g/dL
– Thrombozyten ×109 / L
– Neutrophile ×109 / L
3., Schwere Infektionen
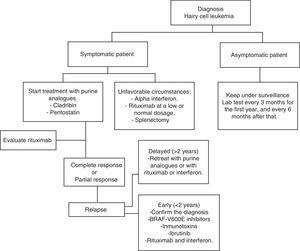
Diagramm für die Behandlung eines Patienten mit Haarzellenleukämie.
Purinanaloga
1960 zeigten die Ergebnisse, dass 30% der Kinder mit schwerem kombinierten Immunschwächesyndrom kein Deaminase-Adenosin-Enzym (ADA) hatten.,40 Darüber hinaus entdeckten sie, dass die Akkumulation der Desoxyadenosintriphosphatform für die Abnahme der Lymphozyten verantwortlich war.Unter Berücksichtigung dieser Beobachtungen wurden Medikamente entwickelt, die irreversibel an ADA binden oder seine Wirkung antagonisieren können. Nach der Behandlung mit Purinanaloga führt die Akkumulation von Desoxyadenosintriphosphat zu einem Bruch und einer Hemmung der DNA-Reparatur, was sich in zellulärer Apoptose niederschlägt.,42
Pentostatin
Auch bekannt als 2′ – Desoxycoformycin (dcF), ein Produkt von Streptomyces-Antibiotika und ADA-Inhibitoren, das erstmals 1980 als erste Purinanaloga zur HCL-Behandlung eingeführt wurde. In Tabelle 3,37,39,43,44 zeigen spezifische Studien die Ansprechraten auf Pentostatin. Die intravenöse Anwendung von Pentostatin im Verhältnis 4 mg/m2 alle zwei Wochen bis zum Erreichen eines vollständigen Ansprechens wurde in den USA zugelassen Pentostatin ist bei Patienten mit Kreatinin-Aufhellung sicher >60mL/min. Eine Dosisreduktion ist jedoch erforderlich, wenn diese Enthärtung zwischen 40 und 60 ml/min liegt.,32 Patienten Hydratation mit 1,5 l intravenöser Lösung mit jedem Pentostatin-Zyklus wird empfohlen.30
Die globale Antwort variiert zwischen 88% und 96%, während die vollständige Antwort zwischen 44% und 81% liegt. Flinn et al. geschätzte Überlebensrate von 5 und 10 Jahren von 90% bzw. 81% mit einer mittleren Follow-up-Dauer von 9,3 Jahren.45 Pentostatin ist im Allgemeinen gut verträglich und die häufigsten Nebenwirkungen sind Anämie, Thrombozytopenie und Neutropenie.,Dennoch wurde berichtet, dass Pentostatin die CD4+ – und CD8+ – Lymphozytenzahl signifikant senkt, was die sekundären Malignitäten und die Inzidenz von Infektionen erhöhen könnte.47,48
Cladribin
Es ist bekannt als 2-chlorodeoxyadenosine (CdA). In Tabelle 4,49-52 zeigen Studien seine Wirksamkeit. Das am häufigsten verwendete Schema besteht aus der Verabreichung von 0,1 mg/kg / Tag in einer kontinuierlichen Infusion für 7 Tage. In einer nicht randomisierten Studie gab es Hinweise darauf, dass es keinen statistisch signifikanten Unterschied in den Ansprechbereichen und Toxizitätsbereichen zwischen Infusionen gab (24h und 2h).,53 Eine weitere randomisierte Studie verglich die tägliche mit der wöchentlichen Cladribin-Verabreichung; Es gab keine signifikanten Ergebnisse in Bezug auf Reaktion, Überleben, globale und Toxizitätsraten.54 Eine andere Studie zeigte, dass das wöchentliche Programm das Infektionsrisiko verringerte.55 Einer der Vorteile der subkutanen Verabreichung ist die Tatsache, dass in den meisten Fällen kein Krankenhausaufenthalt erforderlich ist. 0,14 mg / Tag für 5 Tage hat eine 95% ige Ansprechrate,56 ähnlich der intravenösen Verabreichung. Wöchentliche subkutane Programme haben ähnliche Reaktions-und Toxizitätsraten wie tägliche.,57 Mit nur einem Cladribinzyklus kann eine globale Reaktion von bis zu 100% erzielt werden, und die Gesamtantwortraten unterscheiden sich von 77% bis 95%.49-52 Jehn et al. berichtete ein globales Überleben bei 12 Jahren von 79%.36 Im Allgemeinen ist Cladribin gut verträglich, wobei Zytopenie und Fieber die häufigsten Nebenwirkungen sind.
Studien zur Wirksamkeit von Cladribin in HCL-Fällen.,
| Study | Patients | Follow-up | Plan | % CR | % PR | % GR | Outcome |
|---|---|---|---|---|---|---|---|
| Robak (1999) | 97 | 36 months | 2h infusion IV 0.12mg/kg/d 5 days |
77.3 | 18.,6 | 95.9 | Progression-free survival: 37.4 months |
| Rosenberg (2014) | 83 | NA | Continuous infusion IV 0.1mg/kg/d 7 days |
88 | 12 | 100 | Average global survival: 231 months |
| Goodman (2003) | 207 | 7 years | Continuous infusion IV 0.1mg/kg/d 7 days |
95 | 5 | 100 | Global survival at 108 months: 97% |
| Chadha (2005) | 86 | 9.7 years | Continuous infusion IV 0.,1mg/kg/d 7 days |
79 | 21 | 100 | Global survival at 12 years: 87% |
RC: complete response, RP: partial response, RG: global response.
Until now, there are no randomized prospective studies comparing pentostatin versus cladribine, in part because of the great efficiency of both drugs and due to low HCL incidence., Trotzdem gibt es retrospektive Studien, die belegen, dass beide Medikamente eine ähnliche Wirksamkeit haben, was das vollständige Ansprechen und das Überleben ohne Krankheit betrifft.
Andere Behandlungen
Purinanaloga bleiben die erste Behandlungslinie, aber neue Entdeckungen in Bezug auf die HCL-Pathophysiologie haben zur Schaffung von Arzneimitteln mit unterschiedlichen therapeutischen Zielen geführt. Diese Medikamente sind in der Forschung und einige haben vielversprechende Ergebnisse gezeigt.
Rituximab
Da HCL eine B-Zell-Malignität ist, ist es logisch, einen monoklonalen Antikörper gegen CD20 wie Rituximab zu verwenden., Rituximab wird als eigenständiges Medikament eingesetzt und kann bei Patienten mit einem HCL-Rückfall bei einer Dosierung von 375 mg/m2 einmal wöchentlich für 4-8 Wochen eine Gesamtansprechrate von 10-54% erreichen.58,59 Else et al. retrospektiv wurden 18 Patienten untersucht, die mit Purinanaloga in Kombination mit Rituximab als zweiter Behandlungslinie behandelt wurden, nachdem sie mit Purinanaloga als Einzelwirkstoffen behandelt worden waren. Alle Patienten reagierten mit einer vollständigen Ansprechrate von 89%.60
Rituximab bei 375 mg / m2 pro Woche für 8 Wochen als Ersttherapie nach Verabreichung von 5.,6mg / m2 Cladribin über 2-h IV Infusion für 5 Tage erzeugt eine Gesamtantwort von 100%.61 In besonderen oder ungünstigen Situationen können 100 mg Rituximab pro Woche für 4-6 Wochen verwendet werden. Dies ist weniger teuer und ist in der Regel wirksam, insbesondere wenn es mit Interferon kombiniert wird.
Während Purinanaloga möglicherweise nicht in der Lage sind, HCL zu eliminieren, da die nach Cladribin-Verabreichung festgestellte minimale Restkrankheit (MRD) immer stark CD20+ ist, kann die MRD-Eradikation mit Rituximab erreicht werden. Rivandi et al., in einer Vorstudie wurde nachgewiesen, dass Rituximab in konventionellen Dosen über einen Zeitraum von 8 Wochen mit großer Aktivität wirkt und bei 13 Patienten MRD eliminiert, wenn es 4 Wochen nach der Verabreichung von Cladribin angewendet wird.62
Vemurafenib
Wie bereits beschrieben, ist die BRAF V600E-Mutation der genetische Schlüssel in HCL. Daher ist es ein therapeutisches Ziel, das in den letzten Jahren untersucht wurde. Vemurafenib ist ein oraler Inhibitor von BRAF V600E. Tiacci et al., durchführung einer Studie zur Messung der Vemurafenib-Aktivität und-Sicherheit bei Patienten mit HLC, die nach der Behandlung mit Purinanaloga einen Rückfall erlitten haben oder die auf die Verabreichung von Purinanaloga nicht ansprachen. Die globale Ansprechrate betrug 96% und die vollständige Ansprechrate 35%, mit einem rückfallfreien Überlebensmittel von 19 Monaten. Die Nebenwirkungen waren Hautausschlag, Arthralgie und arthritis.63
Da zwischen der Anwendung von Vemurafenib und dem Auftreten von dermatologischen Malignomen ein positiver Zusammenhang beobachtet wurde, werden häufige Hautuntersuchungen empfohlen.,
Ibrutinib
Ein selektiver und irreversibler Inhibitor der Burton-Tyrosinkinase greift in den B-Zell-Signalweg ein.64 Ein Ibrutinib-klinischer Assay bei Patienten mit HCL-Rückfall hat kürzlich begonnen. Vorläufige Wirksamkeits-und Sicherheitsdaten zeigen Nebenwirkungen wie Eruptionen, Durchfall und Arthralgie. Dieser klinische Test findet derzeit in mehreren Zentren in den USA statt (NCT01841723).
Immunotoxine
Um die monoklonale Antikörper–Zytotoxizität zu erhöhen, wurden Techniken entwickelt, die die Produktion von Antikörper–Toxin-oder Antikörper-Wirkstoff-Konjugaten erleichtern., Ein Immunotoxin ist die Fusion zwischen einem bakteriellen Toxin (d. H. Pseudomonas exotoxin oder Diphtherie) und der variablen Fraktion eines monoklonalen Antikörpers, dessen spezifisches Ziel auf der Oberfläche neoplastischer Zellen wie CD25 oder CD22 gefunden wird. Dieses Toxin wird im Inneren der neoplastischen Zelle freigesetzt und stört die Proteinsynthese.65
BL22 ist ein Immunotoxin gegen CD22, das mit einer abgeschnittenen Form des P. Exotoxins PE38 verschmolzen ist. In einem klinischen Test der Phase II wurde BL22 in 36 HCL-Rezidiv-oder Refraktärerkrankungen getestet., Nach einem Zyklus (40 mg / kg alle zwei Tage, drei Dosen) betrug die vollständige Ansprechrate 25% und die globale Ansprechrate 50%. Diese Reaktionen verbesserten sich auf eine Gesamtansprechrate von 47% und eine globale Ansprechrate von 72% nach der Retreatment (nur bei Patienten mit Zytopenie). Zwei Patienten entwickelten ein urämisches hämolytisches Syndrom, ohne zur Plasmapherese zurückkehren zu müssen.66 Anschließend wurde Moxetumomab Pasudotox als modifizierte Version von BL22 mit höherer Affinität und Zytotoxizität entwickelt., In einem Phase-I-Assay, der 28 Patienten mit HCL-Rückfall und-resistenz umfasste, wurde eine globale Ansprechrate von 86% erhalten, einschließlich einer anhaltenden vollständigen Ansprechrate bei 46% der Patienten.67
Therapeutische Optionen unter ungünstigen Umständen
Obwohl HCL in den meisten entwickelten Ländern mit Cladribin und Pentostatin behandelt wird, ist es eine Tatsache, dass diese Medikamente nicht nur teuer sind, sie sind in Mexiko und in vielen Ländern mit begrenzten Ressourcen nicht verfügbar., Unter diesen Umständen gibt es also andere erschwingliche therapeutische Optionen mit günstigen Ergebnissen
Interferon alpha für die HCL-Patientenbehandlung wurde erstmals 1984 eingeführt. Heute ist seine Verwendung begrenzt, hauptsächlich aufgrund der großen Wirksamkeit von Purinanaloga. Andererseits ist es in Ländern mit geringen wirtschaftlichen Ressourcen eine kostengünstige Option, die ähnliche Ergebnisse wie Cladribin in Bezug auf das globale Überleben erzielt hat. Ruiz-Delgado et al., führte eine Vergleichsstudie zwischen Interferon alpha (n=18) und Cladribin (n=11) durch, bei der der Unterschied im globalen Überleben zwischen beiden Gruppen statistisch nicht signifikant war; 94% nach 217 Monaten in der Interferongruppe und 91% nach 133 Monaten in der Cladribingruppe.68 In einer Studie, die in unserem Zentrum durchgeführt wurde, erhielten neun HCL-Patienten 12 Wochen lang dreimal wöchentlich drei IFN-Mega-Einheiten, anschließend wurden sie 8 Wochen lang bei einer Leukämie-Reaktivierung oder nach 10-monatiger Beobachtung jedes Jahr erneut behandelt., Alle Patienten hatten vor 12 Wochen der Behandlung eine hämatologische Remission. Diese therapeutische Option ist billiger, effektiver und vergleichbar mit anderen Therapieformen mit IFN bei der Behandlung und Aufrechterhaltung von Patienten mit dieser Art von Leukämie.69 Es ist möglich, Interferon mit Rituximab zu kombinieren, ohne die toxischen Wirkungen zu erhöhen und die Wirksamkeit zu verbessern.
Die Splenektomie war die erste Intervention, die das Überleben bei Patienten signifikant veränderte. Heute wird es selten verwendet., Es kann bei Patienten mit schmerzhafter massiver Splenomegalie (>10 cm unter der Rippenkante) und mit minimaler Infiltration des Knochenmarks oder bei Patienten empfohlen werden, die auf eine Behandlung mit Interferon-und Purinanaloga nicht ansprechen.33 Retrospektive Studien zeigen eine vollständige Ansprechrate von 40-62% und eine mittlere Überlebensrate nach 5 Jahren bis zu 68%.70,71 Lad et al. in einer retrospektiven Studie, an der 24 Patienten mit HCL-Diagnose teilnahmen, wurden sie in zwei Gruppen eingeteilt: 17 Patienten erhielten Cladribin und 7 wurden splenektomiert., 75% der Patienten in der Splenektomie-Gruppe zeigten eine totale Remission, 94% in der Cladribin-Gruppe. Ein interessanter Befund beim Vergleich beider Gruppen war, dass keine statistisch signifikanten Unterschiede in Bezug auf das leukämiefreie Überleben und das globale Überleben beobachtet wurden.72
Prognose
Die Überlebenszeit bei Patienten nach der Diagnose betrug 4 Jahre vor Bekanntwerden einer Behandlung aufgrund von Komplikationen aufgrund von Zytopenie, einschließlich Blutungen und Infektionen. Danach gab es bei Splenektomie als Erstbehandlung ein vollständiges Ansprechen von 40-62% und eine Überlebensrate von 5 Jahren bei 61-68%., Dann wurde Alfa Interferon als erstes Medikament mit Vorteilen bei der Behandlung von HCL verwendet. Dennoch war die volle Ansprechrate mit 10% niedrig.73
Heute wird bei Purinanaloga (Pentostatin und Cladribin) ein vollständiges Ansprechen von bis zu 80% der Patienten mit einem medianen Überleben von 10 Jahren induziert. Die globalen Ansprechraten liegen bei 96-100% mit einer vollständigen Ansprechrate von 80% und einer Überlebensrate von 10 Jahren zwischen 85% und 100%.74 Trotzdem versagt ein signifikanter Anteil der Patienten mit HCL in ihrem Ansprechen auf die Behandlung oder wird resistent., Bis zu 48% der Patienten fallen in den folgenden 15 Jahren zurück.75 Die Zukunft von HCL-Patienten ist sehr günstig. Die Herausforderung besteht darin, diese Malignität so früh wie möglich zu identifizieren und mit den verfügbaren Ressourcen richtig zu behandeln.
Interessenkonflikt
Die Autoren haben keine Interessenkonflikte zu erklären.