w poprzednim poście przedstawiliśmy dyrektorów Orto, para i meta w elektrofilowej substytucji aromatycznej. Wcześniej omówiliśmy mechanizm elektrofilowej substytucji aromatycznej i pokazaliśmy, że mechanizm przebiega przez półprodukt karbokalizacyjny.,
dzisiaj połączymy te dwa pojęcia i spróbujemy pokazać, że to, czy podstawnik jest Orto-, para – czy meta – dyrektorem, zależy od tego, w jaki sposób podstawnik wpływa na stabilność pośredniego karbokationu.
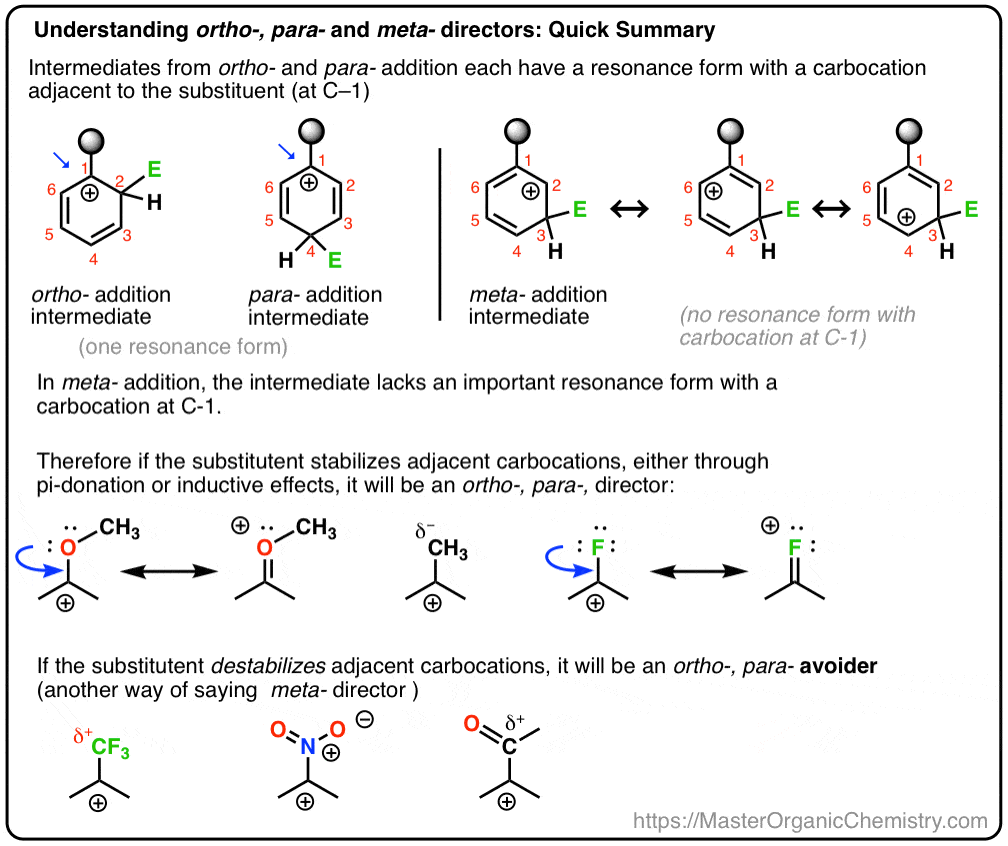
i zobaczymy, dlaczego alternatywna nazwa dla Meta – director może być, „Orto -, para – avoider”. :- )
spis treści
- Karbokacje są stabilizowane przez sąsiadujące Atomy z samotnymi parami
- studium przypadku #1: Orto -, para-Director (OCH3)
- tak, tlen jest Elektronegatywny., Ale Darowizna Jego Samotnej Pary Więcej Niż Nadrabia!
- Diagram reakcji i energii dla dyrektora ortopedycznego
- Case Study #2: a meta – director (CF3).
- to, co nazywamy „Meta– dyrektorami”, jest bardziej jak „Orto -, para– Avoiders”
- Diagram energii reakcji dla Meta – Dyrektora
- więc dlaczego Halogeny są Orto -, Para – dyrektorami?
- Notes
- Quiz Sam!,
- (zaawansowane) Referencje i dalsze czytanie
Karbokacje są stabilizowane przez sąsiadujące Atomy z samotnymi parami
zastanówmy się, które czynniki zwiększają stabilność karbokacji.
Karbokacje są gatunkami ubogimi w elektrony z sześcioma elektronami w swojej powłoce walencyjnej. Tak więc każdy podstawnik, który może przekazać gęstość elektronów w kierunku karbokacji, będzie stabilizowany. Obejmuje to:
- podstawniki uwalniające elektrony, które mogą działać jako „dawcy sigma” (np., wiele grup alkilowych, które przekazują gęstość elektronów poprzez efekty indukcyjne)
- sąsiadujące Atomy z samotnymi parami, które mogą działać jako” donory pi „do karbokacji
- sąsiadujące wiązania C-C pi, które mogą stabilizować karbokację poprzez delokalizację ładunku (innymi słowy” stabilizacja rezonansowa”).
i czynniki destabilizujące karbokacje?
- dowolny podstawnik usuwający gęstość elektronów z karbokationu, np. silne grupy wycofujące elektrony.
więc oto szybki quiz., Biorąc pod uwagę wszystko napisane powyżej, który z dwóch karbokacji poniżej będzie bardziej stabilny?
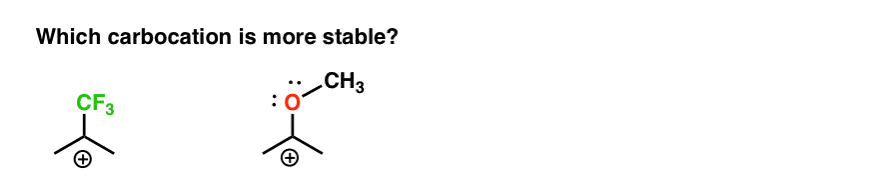
Jeśli potrafisz dobrze odpowiedzieć na to pytanie, jesteś najbardziej na drodze do zrozumienia różnicy między Orto-, para – i meta – reżyserami.
przeanalizujmy dwa studia przypadków. Przyjrzymy się ogólnej elektrofilowej reakcji aromatycznej substytucji benzenu z Orto -, para-director (metoksybenzen) i zbadamy półprodukty, które są otrzymywane z ataku w pozycjach Orto, meta i para., Następnie wykonamy to samo ćwiczenie z meta-reżyserem (trifluorometylobenzenem).
w procesie, mamy nadzieję zrozumieć, dlaczego metoksybenzen sprzyja Orto-i para-produkty, podczas gdy trifluoromethylbenzene sprzyja meta-produkt.
Case Study #1: Orto-,para – Director (OCH3)
Elektrofilowa aromatyczna substytucja metoksybenzenu ma tendencję do dawania Orto – i para – produktów o bardzo małej meta- ., Oto, co dzieje się w nitracji, na przykład:
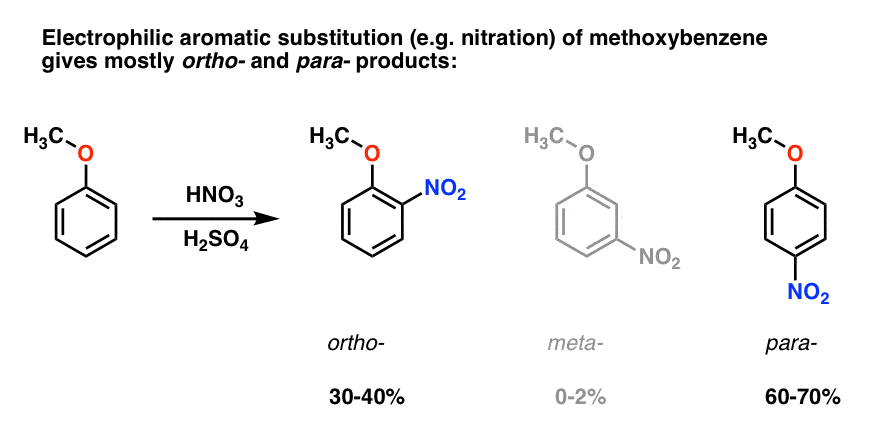
para – produkt dominuje (60-70%), z Orto – blisko (30-40%) i tylko ślad meta– .
Dlaczego?
3. Tak, Tlen Jest Elektronowy. Ale Darowizna Jego Samotnej Pary Więcej Niż Nadrabia!
oto droga prowadząca do Orto – produktu., Zrywamy Wiązanie C–C pi i tworzymy nowe Wiązanie C-E (gdzie E ma reprezentować ogólny elektrofil), generując karbokację stabilizowaną rezonansem:
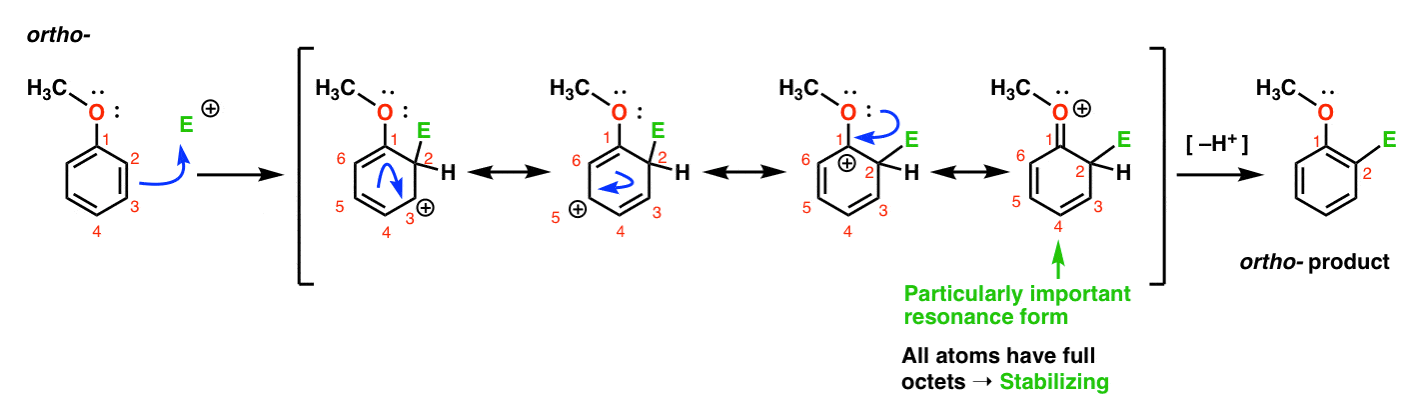
jeśli przyjrzymy się bliżej, należy zauważyć, że możemy utworzyć cztery formy rezonansowe . Trzy z nich posiadają pełne karbokacje (na C1, C3 I C5), ale jest to czwarta – forma rezonansowa, w której występuje Wiązanie C=O pi – co jest szczególnie ważne. Dlaczego? W tej formie rezonansowej wszystkie atomy węgla mają pełny oktet elektronów., To dlatego, że tlen bezpośrednio związany z pierścieniem może przekazać pojedynczą parę do sąsiedniego karbokationu, tworząc wiązanie pi. Jest to przykład darowizny pi.
oto ścieżka ataku elektrofilu na Meta – pozycji. Zauważ różnicę!
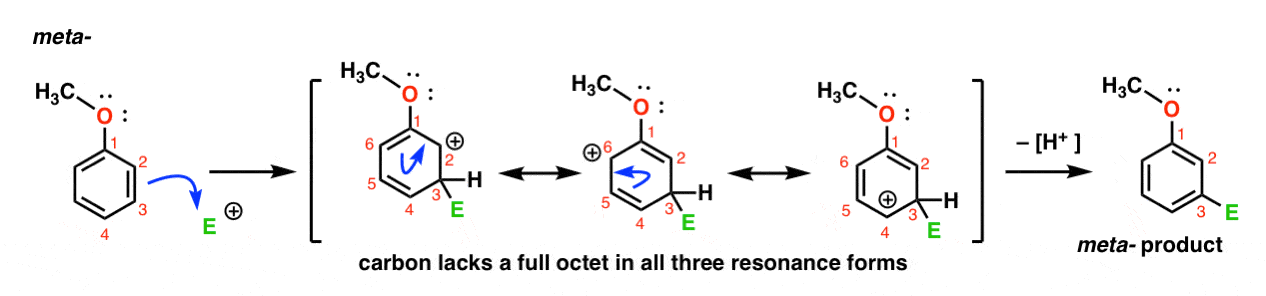
atak elektrofilu w C-3 powoduje karbokalizację, która może być delokalizowana poprzez rezonans do C2, C4 i C6. Nie ma jednak sposobu na „przeniesienie” karbokacji do C1, co oznacza, że nie ma rozsądnej formy rezonansowej, w której wszystkie atomy mają pełne oktety.,
To sprawia, że półprodukt meta – karbokacji jest znacznie mniej stabilny niż półprodukt Orto-karbokacji.
wreszcie, oto reakcja prowadząca do Para – produktu:
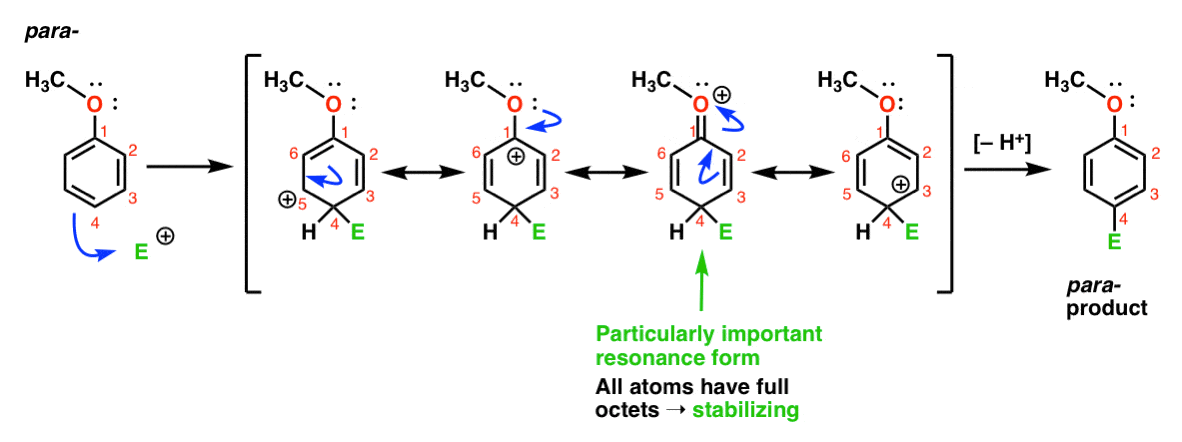
tutaj możemy ponownie narysować formy rezonansowe z karbokacjami na C1, C3 I C5, a także czwartą formę rezonansową, w której dołączony atom tlenu przekazuje parę elektronów do karbokacji na C1, co daje pełny oktet na węglu. Jest to sytuacja zasadniczo taka sama jak w przypadku Orto-pośredniego.,
tak więc, poprzez analizę form rezonansowych (i zapamiętaj moje słowo, możesz zostać poproszony o zrobienie tego samego podczas testu w niedalekiej przyszłości) możemy zobaczyć, że pośrednie karbokacje wynikające z Orto– i para-addycji są znacznie bardziej stabilne niż pośrednie z meta– addycji.
To wyjaśnia, dlaczego dominują Orto – i para-produkty, a meta-produkt jest niewielki.
pomocne może być zwizualizowanie tego poprzez narysowanie diagramu energii reakcji.
4., Diagram energii reakcji dla Orto-para Director
stan przejściowy prowadzący do produktów Orto – i para-addycji jest znacznie niższy w energii niż meta -.
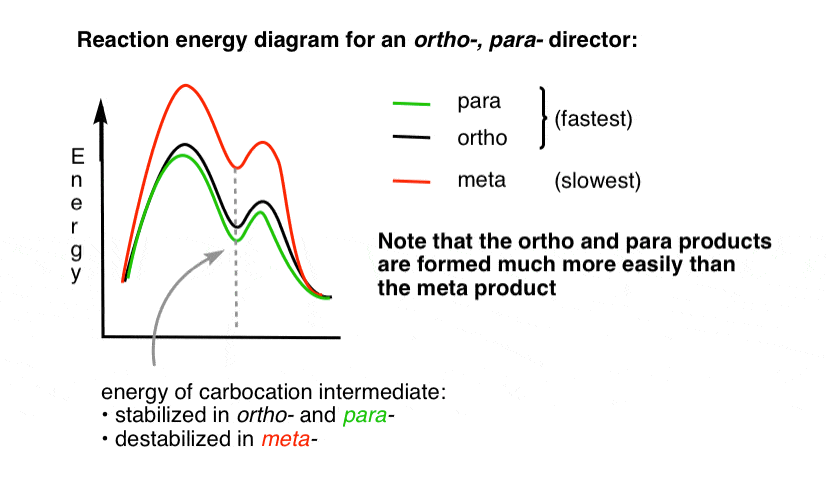
jedno pytanie – dlaczego uważasz, że para – może być preferowana (np. 60% -70% wydajności dla para– produktu nitracji, powyżej, w porównaniu do 30-40% dla Orto -), mimo że istnieją dwie pozycje Orto i tylko jedna para -?
pomyśl o tym przez chwilę, a zajmiemy się tym poniżej.
5. Case Study # 2: a meta-director (CF3).
a co z CF3?, Dlaczego daje meta-produkty? Zróbmy ten sam rodzaj analizy.
oto droga prowadząca do Orto – produktu. Podobnie jak w przypadku Orto-pośredniego powyżej, możemy narysować trzy formy rezonansowe, umieszczając karbokację odpowiednio na C1, C3 I C5.
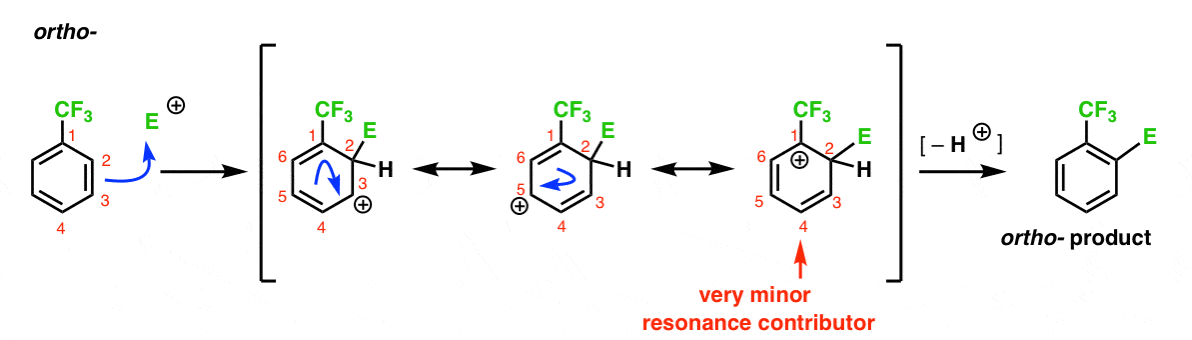
czym się różni w tym przypadku?
różni się obecnością silnej grupy wycofującej elektrony (CF3) w C1, co całkowicie zmienia rozgrywkę.
jak już powiedzieliśmy, karbokacje są destabilizowane przez sąsiadów, którzy wycofują gęstość elektronów., Dlatego spodziewamy się, że będzie to bardzo niewielki udział rezonansu, w wyniku czego ładunek dodatni jest delokalizowany tylko nad C3 I C5.
zauważyliśmy, że CF3 jest meta – dyrektorem. Rozsądnie jest myśleć, że jest coś w Meta-co czyni go szczególnie stabilnym. Analizując pośredni dla meta-produktu poniżej, możesz zobaczyć dlaczego?
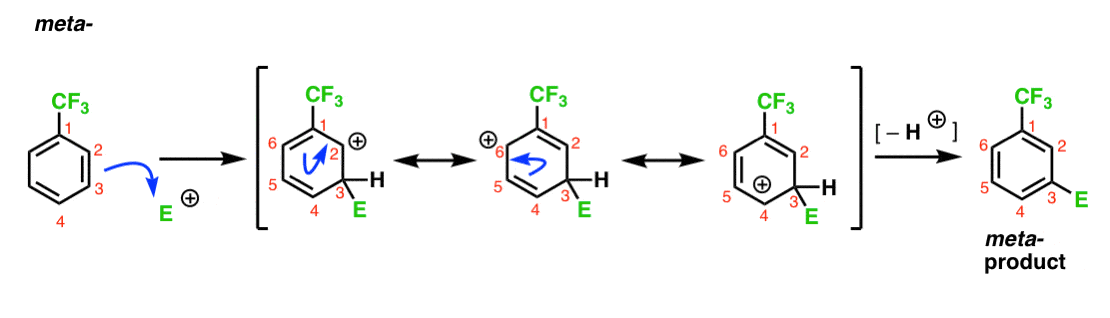
Hmmm. Nie ma półproduktu, w którym węgiel ma pełny oktet. Nie ma szczególnie stabilnych form rezonansowych.
to coś w rodzaju „meh”, prawda?
6., To, co nazywamy „Meta-reżyserami”, jest bardziej jak”Orto -, para– Avoiders”
z drugiej strony, nie ma też żadnych szczególnie niestabilnych form rezonansowych. Ponieważ ładunek dodatni znajduje się na C2, C4 i C6, unika się sytuacji, w której ładunek dodatni sąsiaduje bezpośrednio z grupą CF3 wycofującą elektrony. W ten sposób ładunek dodatni ulega delokalizacji w całym pierścieniu, w przeciwieństwie do sytuacji ortodoksyjnej, powyżej.
więc jest to „mniej zły” pośrednik niż Orto-.,
wreszcie, para – intermediate ma ten sam problem co Orto- ; pośredni z dodatnim ładunkiem na C1, sąsiadujący z CF3.
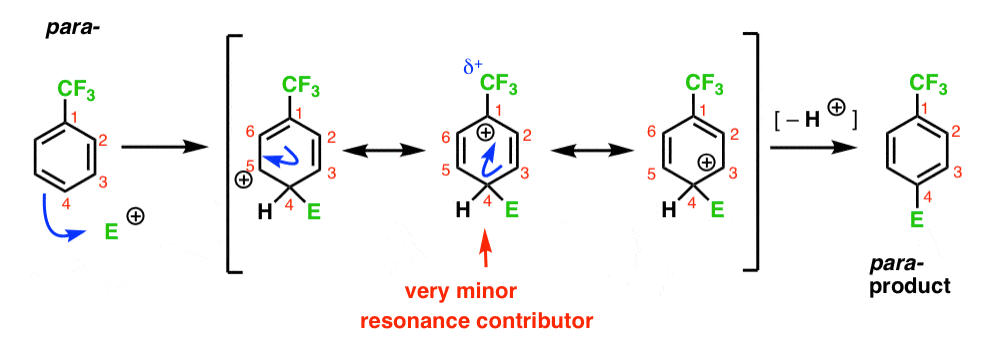
powoduje to tylko dwie ważne formy rezonansu, co prowadzi do mniej zdelokalizowanego (a zatem mniej stabilnego) pośredniego karbokalizacji.
Reasumując: meta carbocation intermediate jest bardziej stabilny niż Orto– lub para – intermediates.
ale nie dlatego, że sam substytut ma jakikolwiek stabilizujący wpływ na Meta– pośredni. .,
uważam, że bardziej pomocne jest myślenie o meta – jako o mniej niestabilnym niż Orto – i para– .
w ten sposób nie jest tak bardzo, że CF3 jest meta – reżyserem, tak bardzo , że jest „Orto–, para– avoiderem. „
7. Diagram energii reakcji dla Meta-Dyrektora
oto szkic diagramu energii reakcji:
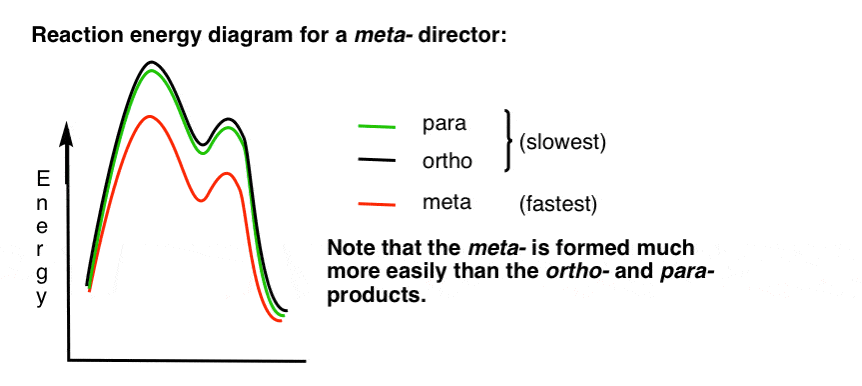
w porządku.
powrót do naszego pierwotnego pytania. Która karbokacja jest bardziej stabilna?,
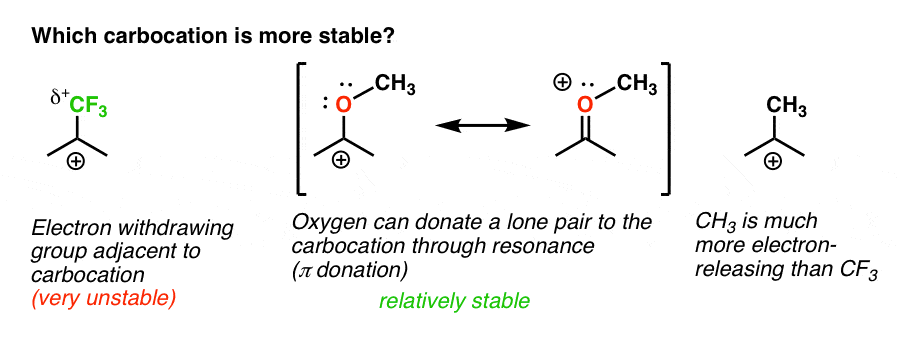
wyraźnie, karbokacja z sąsiednim tlenem noszącym samotną parę jest znacznie bardziej stabilna niż karbokacja przylegająca do grupy wycofującej elektrony, takiej jak CF3.
i na tym polega różnica między Orto-, para – director jak OCH3 i meta – director jak CF3.
warto zauważyć, że większość grup alkilowych (np. CH3 ) nie ma samotnej pary, ale nadal są Orto -, para – dyrektorami., Jest to zgodne ze wszystkim, czego dowiedzieliśmy się wcześniej o tym, jak grupy alkilowe generalnie stabilizują się dla karbokacji-przypomnijmy, że stabilność karbokacji na ogół wzrasta wraz z substytucją
więc o tych para– produktach…
Dlaczego para – produkty wytwarzane są w większym tempie niż Orto- ?
odpowiedź jest bezpłodna.
atak na pozycji para jest mniej obciążony przez podstawnik niż atak na pozycji Orto.
więc dlaczego Halogeny są Orto -, Para-dyrektorami?
To pozostawia nam nieco trudny przykład halogenów.,
Dlaczego fluor, chlor, brom i jod są Orto -, para -, mimo że są grupami dezaktywującymi?
odpowiedź nie powinna być zaskoczeniem, jeśli podążasz za nią, ale zostawimy to do następnego posta, ponieważ ten już jest wystarczająco długi.
następny wpis: dlaczego halogeny są ortopedyczne?
uwagi
Uwaga 1. Bardziej poprawne jest stwierdzenie, że dominują Orto-i para-produkty, ponieważ stany przejściowe prowadzące do tych produktów są niższe w energii, a nie Energie samych półproduktów., Przecież to energia stanów przejściowych decyduje o barierze aktywacji, a zatem o szybkości reakcji.
Uwaga 2. Lub hiperkonjugacji, której większość podręczników (z zauważalnym wyjątkiem Maitlanda Jonesa) na ogół unika.
Quiz Sam!,/div>
 i nacisnąć, aby odwrócić
i nacisnąć, aby odwrócić
i nacisnąć, aby odwrócić  i naciśnij, aby włączyć
i naciśnij, aby włączyć 
(zaawansowane) linki i dodatkowe literatura
- A., F. Holleman, Die direkte Einführung von Substituenten in den Benzolkern
Rec. Trav. Chim. Pays-Bas 1910, 12, 455-456
DOI: 10.1002/recl.19100291205
A. F Holleman z 1910 stwierdził, że orientacja ortopedyczna jest związana z aktywacją, a orientacja meta-z dezaktywacją. - niektóre obserwacje dotyczące utrudnień sterylnych i wpływu podstawników na stosunek Orto: para w substytucji aromatycznej
P. B. D. de la Mare
J. Chem. Soc. 1949, 2871-2874
doi: 10.1039/JR9490002871
w artykule de la Mare omawia różne czynniki, które mogą tłumaczyć przewagę para nad selektywnością Orto, po podzieleniu wydajności produktu Orto przez 2 (ponieważ istnieją 2 pozycje Orto, ale tylko 1 pozycja para w monosubpodstawionych benzenach)., - wpływ objętościowy grup alkilowych w związkach aromatycznych. Część V. monosulfonacja p-cymene
R. J. W. Le Fèvre
J. Chem. Soc. 1934, 1501-1502
doi: 10.1039/JR9340001501
W P-cymenie głównym produktem otrzymywanym w wyniku sulfonowania elektrofilowego jest 2-Produkt (Orto do grupy metylowej), prawdopodobnie ze względu na stery. - Effects of Alkyl Groups in Electrophilic Additions and Substitutions
Cohn, H., Hughes, E., JONES, M. and PEELING, M. G.
Nature 1952, 169, 291
Doi: 1038/169291a0
w artykule przedstawiono dane porównujące azotowanie t-butylobenzenu i toluenu., T-butylobenzen jest znacznie bardziej P-kierujący niż toluen (79,5% para dla T-butylobenzenu vs 40% para dla toluenu), co jest prawdopodobnie spowodowane sterycami (podejście Orto jest blokowane przez większą grupę t-butylową). - rozkład izomerów w Mononitracji etylo-i Izopropylobenzenu. Further Evidence for a Steric Effect in Isomer Distribution
Herbert C. Brown and W. Hallam Bonner
Journal of the American Chemical Society 1954, 76 (2), 605-606
DOI: 10.,1021 / ja01631a084
tabela II w niniejszym artykule ilustruje, że produkt Orto otrzymywany z azotowania monoalkilobenzenów zmniejsza się wraz ze wzrostem grupy alkilowej (np. t-butylobenzen daje bardzo mało produktu Orto po azotowaniu w porównaniu z toluenem). - anomalna reaktywność Fluorobenzenu w elektrofilowej substytucji aromatycznej i związanych z nią zjawiskach
Joel Rosenthal i David I., Schuster
Journal of Chemical Education 2003, 80 (6), 679
Doi: 1021/ed080p679
bardzo ciekawy artykuł, nadaje się do ciekawych licencjatów, i omawia coś, co większość praktykujących chemików organicznych będzie wiedzieć empirycznie-fluorobenzen jest prawie tak reaktywny jak benzen w EAS lub Friedel-Crafts reakcji, co jest sprzeczne z intuicją, gdy weźmie się pod uwagę efekty elektroniczne! - Chemia organiczna
N. Haworth, C. K. Ingold, T. A. Henry
Ann. REP.Prog. Chem. 1926, 23, 74-185
DOI: 10.1039 / AR9262300074
wczesna praca omawiająca efekty reżyserskie w EAS. Pp., 136 i 137 zawierają liczby, które przedstawiają przepływ elektronów. Pg. 140 omawia meta-kierunek, który występuje z „przeciwnych” powodów jako kierunek o / p przez grupy alkilowe.
i — – charakter efektu przemiennego w łańcuchach węglowych. Część XXIII. Anomalous orientation by halogens, and its bearing on the problem of the Orto–para ratio, in aromatic substitution
Christopher Kelk Ingold and Charles Cyril Norrey Vass
J. Chem. Soc. 1928, 417-425
DOI: 10.,1039 / JR9280000417
w artykule omówiono efekty kierowania w 1,2-dihalobenzenach.
i — – charakter efektu przemiennego w łańcuchach węglowych. Part XXII.an attempt further to define the probable mechanism of orientation in aromatic substitution
Christopher Kelk Ingold and Florence Ruth Shaw
J. Chem. Soc. 1927, 2918-2926
DOI: 10.,1039 / JR9270002918
wczesna praca wpływowego chemika organicznego, Prof. C. K. Ingolda, stwierdzająca, że halogenobenzeny indukcyjnie wycofują elektrony, ale jednocześnie stabilizują rezonans.